

In cleanrooms, GMP compliance ensures consistent quality and safety by requiring detailed monitoring and accurate data records. For cultivated meat facilities, this is particularly important, as even minor deviations in cleanroom conditions can compromise cell growth or contaminate production batches.
Key takeaways:
- GMP Standards: Focus on data integrity, following the ALCOA+ framework (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available).
- Critical Parameters: Monitor air particles, microbial counts, temperature, humidity, and pressure to detect risks early. This requires selecting precise sensors capable of maintaining these critical parameters.
- Data Systems: Use validated bioprocess control systems with role-based access, audit trails, and secure storage for both electronic and paper records.
- Common Risks: Avoid errors in manual data handling, configuration changes without control, and improper storage practices.
- Tailored GMP for Cultivated Meat: Adjust monitoring strategies to address unique risks like bioreactor conditions and cleaning agent residues.
For cultivated meat R&D, robust data management ensures product safety, regulatory compliance, and scalable operations. Address known vulnerabilities proactively to avoid costly regulatory issues later.
Key GMP Requirements for Cleanroom Data Integrity
Understanding ALCOA+ Principles
The cornerstone of GMP data integrity lies in the ALCOA+ framework. Regulatory bodies like the MHRA, EMA, and WHO use it to determine whether cleanroom records can be trusted. ALCOA+ stands for: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available. Each of these terms carries practical importance in cleanroom operations.
- Attributable: Every entry - whether it's a particle count, pressure reading, or cleaning log - must clearly show who recorded it, along with the date, time, and relevant instrument details.
- Legible: Records must be easy to read and decipher, ensuring clarity during reviews or inspections.
- Contemporaneous: Data must be recorded in real time. Delayed or retrospective entries can compromise the reliability of records.
- Original: Data should remain in its first-captured form, without unauthorised edits or alterations.
- Accurate: Recorded values must truly reflect the observed results, free from errors or manipulation.
- Complete: All relevant entries, including deviations or out-of-specification results, must be documented.
- Consistent, Enduring, and Available: Records should follow the correct sequence, be preserved intact for the required retention period, and be readily accessible for review or inspection.
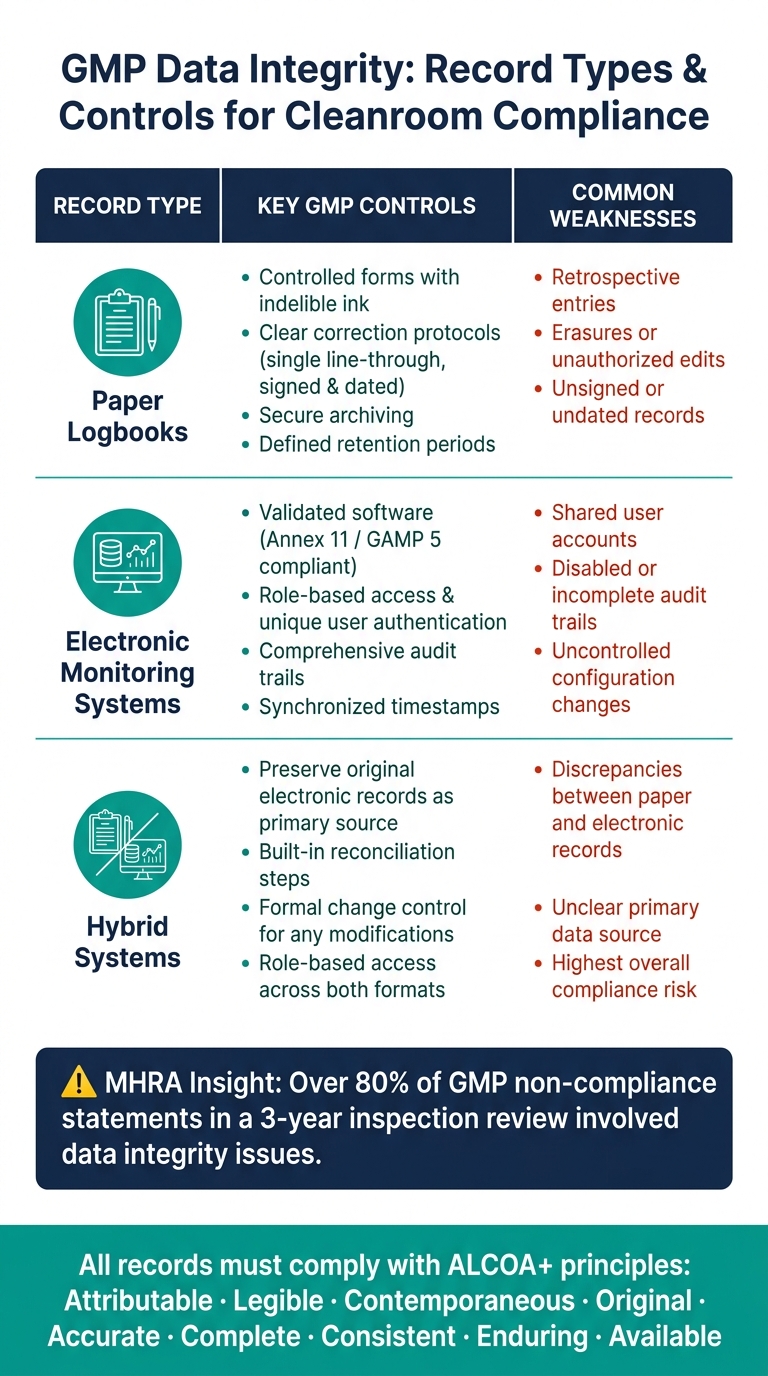
Regulators place a strong emphasis on these principles. For instance, an MHRA inspection review revealed that over 80% of GMP non-compliance statements in a three-year span involved data integrity issues [5]. To embed ALCOA+ into daily workflows, facilities can adopt well-structured forms, enforce mandatory fields, and conduct regular audit trail reviews.
With ALCOA+ as the foundation, the next step is to ensure these principles are upheld across paper, electronic, and hybrid systems.
Ensuring Data Integrity Across Formats
In cultivated meat facilities, where data directly influences batch release decisions, maintaining integrity across all record formats is non-negotiable. GMP requires the same level of integrity for both paper and electronic records, though the specific controls may differ depending on the format.
- Paper systems: Best practices include using controlled forms with indelible ink and clear correction protocols (e.g., single line-through corrections with signatures and dates). Secure archiving and adherence to defined retention periods are also critical.
- Electronic systems: These should operate on validated software that complies with Annex 11 and GAMP 5. Key features include role-based access, unique user authentication, comprehensive audit trails, and synchronised timestamps. Regular audit trail reviews are essential to identify and address any irregularities.
- Hybrid systems: These represent the highest risk since they involve both electronic and paper records. For example, when an instrument generates electronic data that is later transcribed into a paper log, the original electronic output must be preserved as the primary record. Reconciliation steps should be built into the workflow to detect and resolve any discrepancies between the electronic and paper records. This is particularly critical in cultivated meat production, where even minor data inconsistencies could compromise contamination control measures.
The table below summarises the key controls and common weaknesses for each record type:
| Record Type | Key GMP Controls | Common Weaknesses |
|---|---|---|
| Paper logbooks | Controlled forms, indelible ink, clear correction protocols, signed and dated entries | Retrospective entries, erasures, unsigned records |
| Electronic monitoring systems | Validated software, role-based access, audit trails, time synchronisation | Shared user accounts, disabled or incomplete audit trails |
| Hybrid systems | Preserve original electronic records; implement reconciliation steps | Discrepancies between paper and electronic records, unclear primary data source |
To ensure compliance, records should be categorised by their criticality. For cultivated meat facilities, data tied to batch release decisions or contamination control (e.g., environmental monitoring results, HVAC alarm logs, or filter integrity test data) should be subject to the strictest access controls, frequent reviews, and robust audit trail management.
sbb-itb-ffee270
GMP Cleanroom Routine Environmental Monitoring & 21CFR part 11 Data Integrity
Managing the Cleanroom Data Lifecycle
GMP Data Integrity: Record Types & Controls for Cleanroom Compliance
Stages of the Cleanroom Data Lifecycle
The cleanroom data lifecycle in cultivated meat facilities involves several stages, each with specific compliance requirements.
Data generation marks the beginning. This includes readings from instruments like particle counters, differential pressure sensors, temperature and humidity probes, viable air samplers, surface contact plates, and cleaning verification logs. For each parameter, there must be a documented sampling frequency, a designated operator, and a calibrated instrument. Aligning these monitoring tasks with production stages - such as inoculation, cell expansion, or harvest - helps demonstrate how environmental control links directly to product quality and safety.
Once generated, data enters the capture and transfer stage. Ideally, electronic systems should automatically record readings with time-stamped entries tied to individual user accounts. For paper-based entries, data must be logged in real time using indelible ink, with reconciliation checks in place when transferring the data to electronic systems.
The storage phase is equally critical. Both raw and processed data must be preserved so that any reported value can be traced back to its original record. This requires secure, validated repositories with role-based access controls and regular backup testing. Backups should be stored in a separate location from the primary system and periodically verified to ensure they can be restored when needed.
Finally, archiving concludes the lifecycle. Records transition to a read-only, controlled-access state once they are no longer actively used but must remain retrievable for the required retention period. In cultivated meat facilities, archiving development-phase data can also support future validation efforts.
A clear understanding of these stages is essential to managing risks effectively, as outlined below.
Common Risks in Data Management
Data handling during transfers poses considerable risks. Manual transcription errors and retrospective entries can undermine data integrity. To avoid this, all entries must adhere to ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, plus Completeness, Consistency, Enduring, and Available) in real time.
Configuration changes are another major concern. Adjustments to alarm limits, sensor mappings, or system clock settings without formal change control can compromise the reliability of data recorded before and after the change. Additionally, storage failures - whether due to corrupted databases, untested backups, or paper archives damaged by environmental factors - can make critical records inaccessible. To mitigate these risks, ensure every data stream is mapped to a designated archive point with clear ownership, reducing the likelihood of vulnerabilities being flagged during regulatory inspections.
GMP Controls for Cleanroom Monitoring Systems
Critical Controls for Monitoring Systems
A monitoring system that meets GMP standards relies on calibrated sensors, secure data handling, and effective alarm management. Sensors for parameters like temperature, relative humidity, differential pressure, non-viable particle counts, and viable microbial sampling must be calibrated according to documented schedules and traceable to recognised standards. Automating data transfer from these sensors, complete with synchronised time stamps, minimises the risk of manual errors.
Alarm management is equally crucial. Alarm limits should align with regulatory frameworks such as ISO 14644-1 class limits and EU GMP Annex 1 guidance. Every alarm triggered must be accompanied by a recorded response, including user details, time stamps, and any comments. Failing to document an alarm response creates a compliance vulnerability.
Role-based access controls must be strictly enforced across the system. Administrator-level authorisation should be required for any changes to alarm limits, sensor configurations, or system clock settings, and these changes must follow a formal change control process. Audit trails are mandatory for all GMP-related actions, such as configuration updates, data deletions, electronic signatures, and sensor adjustments. These trails need regular reviews, as outlined in MHRA data integrity guidance and EU GMP Annex 11.
For cultivated meat production systems, these controls are particularly important, as environmental conditions have a direct impact on cell viability.
Once these controls are in place, the system must be validated and changes carefully managed to ensure ongoing compliance.
Validation and Change Control Procedures
Monitoring systems must undergo validation through IQ, OQ, and PQ stages to verify sensor accuracy, alarm functionality, data integrity, backup processes, and audit trails. Alternatively, a lifecycle approach aligned with GAMP 5 principles and Annex 11 can be used.
EU GMP Annex 1 (2022 revision) requires environmental monitoring systems to be "suitably qualified and validated" and mandates that electronic records meet Annex 11 standards for integrity, security, and traceability. These requirements set the baseline for any GMP-compliant facility.
Any modifications that could affect data integrity, alarm functionality, or traceability must go through a formal change control process. Even seemingly minor updates, such as software patches, can disrupt audit trails and should not be implemented without a prior impact assessment.
Different types of data require tailored GMP controls to ensure accurate and timely reporting.
Data Types and Compliance Requirements Compared
Each data type in cleanroom monitoring has specific requirements to uphold GMP compliance. The table below outlines the key controls for various data types:
| Data type | Monitoring mode | Key GMP controls | Limit basis |
|---|---|---|---|
| Non-viable particle counts | Continuous or frequent during operations in Grade A/B; routine in other grades | Validated particle counters; automatic data capture; alarmed; calibration traceable to standards; audit trail for configuration changes | ISO 14644-1 class limits; Annex 1 guidance for Grade A/B zones |
| Differential pressure | Continuous; alarmed | Calibrated pressure transmitters; automatic, time-stamped recording; logged alarm acknowledgements; maintain 10–15 Pa differential between zones | Annex 1; facility-specific room classification design |
| Temperature and relative humidity | Continuous for critical processes; periodic elsewhere | Calibrated probes; automatic data capture; trend analysis; alarm limits based on process and regulatory needs | Process knowledge; regulatory guidance; product sensitivity |
| Viable airborne microbes | Intermittent (active air sampling); increased frequency for critical operations | Qualified samplers; controlled sampling procedures; chain-of-custody to laboratory; results linked to batch and location; investigation-ready records | EU GMP Annex 1 microbial limits by grade |
| Surface contact results | Periodic; post-cleaning and post-operation | Controlled sampling methods; laboratory traceability; results reviewed against grade-specific limits; linked to cleaning records | EU GMP Annex 1; facility SOPs |
Each data type requires defined acceptance criteria, regular review schedules, retention policies, and an escalation process for deviations. Applying uniform review standards across all data types is a common mistake that regulators are increasingly scrutinising. Tailoring the review process to the specific needs of each data type ensures compliance and operational efficiency.
Reporting, Review, and Corrective Actions
Creating Compliant Reports
Reports aligned with ALCOA+ standards must be thorough, precise, and accessible for audits. A GMP-compliant cleanroom monitoring report should be concise, verifiable, and capable of supporting batch release decisions while demonstrating environmental control. At a minimum, these reports should:
- Cover the monitoring period and scope.
- Summarise sampling activities compared to the planned schedule.
- Clearly state whether any alert or action limits were exceeded.
Trend analysis is a key component of these reports, employing statistical tools like control charts, moving averages, and excursion rates per 100 samples to identify gradual changes. For instance, a monthly trend showing a steady increase in viable counts near a bioreactor harvest line provides far more insight than a single out-of-limit event. Adding annotations, such as maintenance activities, process adjustments, or personnel changes, makes the data easier to interpret and more audit-ready.
Audit trail reviews are another critical step, requiring trained personnel to document their findings meticulously. This includes recording who reviewed specific system events, noting any anomalies, and detailing follow-up actions, all within a signed and dated record.
Report frequency should align with the associated risk. For example:
- Batch-associated reports are created for each production run.
- Routine environmental monitoring summaries are typically weekly or monthly.
- Trend reports are prepared monthly or quarterly to identify early signs of drift.
The chosen reporting frequency must be justified in standard operating procedures (SOPs) and consistently adhered to. These protocols also provide the foundation for initiating corrective actions when deviations are identified.
Addressing Compliance Failures
When deviations occur, a structured, traceable response is essential. Each deviation should have a unique identifier, a clear description, and a risk assessment that evaluates both product impact and data integrity. Deviations must also be classified (minor, major, or critical) and linked to specific batches or production runs to assess whether batch release is affected or additional testing is needed.
The CAPA (Corrective and Preventive Action) framework is central to addressing GMP failures. Effective CAPA requires more than attributing events to "human error." EMA and PIC/S guidance emphasises:
"failure to adequately investigate critical deviations, OOS results and data integrity issues" is a recurrent cause of enforcement actions.
Root cause analysis tools, such as the 5 Whys or fishbone diagrams, are invaluable for uncovering systemic issues - whether these relate to procedural gaps, insufficient training, or weaknesses in technical controls. Corrective actions should address the risks identified during data capture and storage.
Each CAPA must include measurable effectiveness criteria. For example: "no repeated Grade B viable excursions above the action level for six months." Additionally, follow-up reviews are essential to ensure these criteria are met. Common CAPA metrics include:
- The number of open actions.
- Average time to closure.
- Percentage of actions completed on time.
- Rate of repeat deviations, which serves as a strong indicator of CAPA effectiveness.
A review of GMP warning letters from 2015 to 2019 revealed that 65–70% of data integrity citations stemmed from inadequate investigations, missing documentation, or failures to review and report data properly [2]. This highlights the importance of robust reporting and a responsive CAPA framework as evidence of a well-controlled facility.
Maintaining GMP Compliance in Cultivated Meat Facilities
To ensure safety and quality in cultivated meat production, facilities must adapt established GMP controls to meet the specific challenges of this emerging field. Since cleanroom data integrity plays a critical role in maintaining product safety, refining GMP practices for cultivated meat is essential.
Tailoring GMP Practices for Cultivated Meat
GMP frameworks like EU Annex 1, originally created for pharmaceuticals, require adjustments to address the unique risks in cultivated meat production. A formal risk assessment, such as an FMEA or HACCP-style analysis, provides a solid foundation for aligning GMP principles with each stage of production. Critical operations like cell bank thawing, bioreactor inoculation, cell expansion, and harvest demand appropriate cleanroom classifications, gowning protocols, and environmental monitoring as specified in Annex 1. Meanwhile, downstream tasks like scaffold handling and packaging can adhere to food-grade hygiene GMP standards under Regulation (EC) No 852/2004, provided traceability and data integrity remain intact throughout the process [6][9][14].
Environmental monitoring strategies should focus on organisms relevant to cultivated meat and food safety, rather than solely targeting traditional pharmaceutical pathogens. Sampling should be prioritised in high-risk areas, such as those near open bioreactors, media preparation zones, and scaffold handling stations. These locations should be selected based on documented airflow patterns and personnel movement analyses [9][10].
Given the high volume of data generated by cultivated meat bioreactors, systems must be capable of capturing, time-stamping, and securely storing this data in a validated repository. The original raw data file from the instrument should always be identified as the primary record to ensure compliance [7][8].
Cleaning and disinfection protocols also require careful consideration. Residues deemed acceptable in pharmaceutical settings could interfere with cell adhesion or differentiation in cultivated meat production. Verification data for cleaning agents should be collected and maintained as part of the environmental monitoring programme [3][4].
Using Industry Resources Like Cellbase
Specialised procurement platforms are invaluable for meeting the specific needs of cultivated meat facilities. GMP-compatible cleanroom equipment should meet ingress protection and cleanability standards, while also integrating seamlessly with validated data systems. Suppliers must provide detailed specifications, including audit trail capabilities, data export formats, alarm configurations, and calibration procedures, alongside the equipment.
Cellbase, the first B2B marketplace tailored exclusively to the cultivated meat industry, addresses these requirements. With a focus on cultivated meat applications, Cellbase simplifies the procurement process for bioreactors, sensors, environmental monitoring systems, and other infrastructure designed for these specialised environments. The platform ensures that equipment is compatible with the data outputs required for monitoring and compliance [4].
When sourcing systems through Cellbase, consider the following criteria to ensure they meet GMP requirements:
- Data integrity features: secure audit trails, time-stamped records, role-based permissions, and unique user logins
- System compatibility: support for standard communication protocols and APIs for centralised data storage
- Calibration and maintenance: availability of comprehensive documentation
- Qualification support: supplier-provided IQ/OQ templates for streamlined validation
- Cleanroom suitability: materials and designs that facilitate cleaning
Requesting supplier documentation early in the procurement process can help avoid potential qualification issues down the line [3][4].
Conclusion
Key Points Recap
GMP compliance in cleanroom data management revolves around demonstrating control - over processes, records, and the decisions informed by that data. If a record lacks reliability, the process it documents becomes equally questionable. This principle applies universally, whether dealing with environmental monitoring logs, bioreactor outputs, deviation reports, or calibration certificates.
Four central themes have emerged throughout this discussion. First, data integrity, guided by ALCOA+ principles, is the cornerstone of compliant cleanroom documentation. Second, lifecycle management ensures data is recorded accurately, reviewed promptly, stored securely, and retained for the required duration. Third, validated and change-controlled monitoring systems form the technical foundation that no SOP can replace. As highlighted by the MHRA’s analysis of GMP inspections from 2016 to 2021, common deficiencies continue to include incomplete records and insufficient audit trail reviews [1]. Finally, accurate and traceable reporting ensures raw data can be linked to batch decisions, investigations, and corrective actions, meeting regulatory expectations.
For cultivated meat facilities, these principles take on even greater importance. The challenge of combining R&D-style workflows with production-level controls demands robust data governance to bridge both operational environments. Proper cleanroom data management not only ensures consistency and reproducibility but also prepares facilities for scale-up while demonstrating product safety to regulators, investors, and consumers.
The most actionable advice? Address known risks before auditors highlight them. Vulnerabilities such as hybrid paper–electronic systems, shared user logins, delayed data reviews, and uncontrolled local storage are predictable and preventable. Proactively resolving these issues is far more effective - and less costly - than reconstructing a data trail after a quality incident.
For teams seeking monitoring equipment, sensors, or infrastructure tailored to these needs, Cellbase provides a targeted solution. This marketplace is designed specifically for the cultivated meat sector, connecting buyers with suppliers experienced in GMP-compliant environments.
FAQs
How can ALCOA+ be demonstrated in daily cleanroom records?
To apply ALCOA+ principles in daily cleanroom records, ensure the following:
- Attributable: Clearly identify the person responsible, including timestamps for each entry.
- Legible: Records must be easy to read and free from ambiguity.
- Contemporaneous: Document information at the time the activity occurs.
- Original: Retain the first recording of data, not copies or transcriptions.
- Accurate: Ensure all entries reflect the true data without errors.
- Complete: Include all relevant data and metadata without omissions.
- Consistent: Maintain a logical, sequential order in the records.
- Enduring: Use formats and materials suitable for long-term preservation.
- Available: Keep records accessible for review or audits when needed.
These steps are critical for ensuring compliance with Good Manufacturing Practice (GMP) in cleanroom data management.
What are the main data integrity risks in hybrid paper–electronic systems?
Storing information across multiple locations introduces complications in verifying data accuracy. Additionally, manual data entry increases the risk of human errors, while poorly controlled or standalone systems leave records vulnerable to manipulation or deletion. These issues underscore the need for strong data management practices to uphold compliance and preserve data integrity.
What evidence do inspectors expect for monitoring system validation and change control?
Inspectors often ask for documented evidence that shows system validation. This includes testing critical parameters like:
- HEPA filter integrity: Ensuring the filters meet required performance standards.
- Airflow and pressure differentials: Verifying that these are within acceptable ranges to maintain controlled environments.
- Environmental monitoring data: Demonstrating that the facility meets cleanliness and contamination control requirements.
Beyond validation testing, maintaining records of change control activities is equally important. This covers actions like filter replacements or facility modifications, which help prove that the system continues to perform as expected and complies with regulatory standards.









