

如果先编辑后检查,您可以将偏离目标的更改修正到克隆中。 我会保持工作流程简单:选择风险最低的编辑方法,保持编辑器曝光时间短,然后在发布前测试预测的偏离目标位点和克隆稳定性。
对于生物工艺工程师、细胞培养科学家和培养肉类研发团队来说,重点很简单。CRISPR系统仍然可以在近似匹配位点进行切割,通常可以容忍3–6个错配,这些错误可能会传递到扩展的单细胞克隆中。文章将风险控制分为三个阶段:编辑前 , 编辑中, 和编辑后.
以下是完整的清单,用简单的术语表达:
-
选择风险最低的编辑工具
- 在不需要双链断裂的情况下,使用碱基编辑或引物编辑
- 如果只需要基因调控,使用 dCas9-基础调控
- 如果需要核酸酶,请从 高保真Cas9变体开始
-
锁定起始材料
- 确认细胞系身份
- 检查支原体
- 记录传代次数
- 在工作系中测序实际目标位点,而不仅仅是参考基因组
-
在湿实验前筛选指南
- 使用基于比对和基于评分的脱靶工具一起
- 优先选择脱靶特征更干净的引导,而不是仅仅具有更高靶向活性的引导
- 注意引导长度,40–60% GC含量, 和同聚物运行
-
限制细胞内的暴露
- 尽可能使用RNP或mRNA递送,而不是质粒或病毒系统
- 使用最低有效剂量
- 避免仅仅为了强制转染结果而延长编辑器的持久性
-
为高风险情况添加额外的控制
- 考虑配对核酸酶
- 在时间重要时,使用可诱导的, 分裂Cas9, 或光控系统
- 在需要时添加抗CRISPR蛋白 作为关闭步骤
-
编辑后正确验证
- 首先确认目标编辑
- 使用靶向扩增子NGS检查每个预测的非目标位点
- 当项目风险较高时,转向GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, 或WGS
- 对于碱基或 引导编辑器, 在相关情况下添加RNA水平检查
-
不要仅凭序列释放单个克隆
- 比较2–3个独立克隆
- 使用未编辑的亲本对照
- 去除具有不稳定性或表型漂移的克隆
- 仅在编辑状态、非目标筛查和记录全部完成时释放
一个简短的思考方式:设计以避免非目标切割,交付以限制在细胞内的时间,然后根据项目风险的深度进行验证. 贯穿整个作品的线索就是这个。
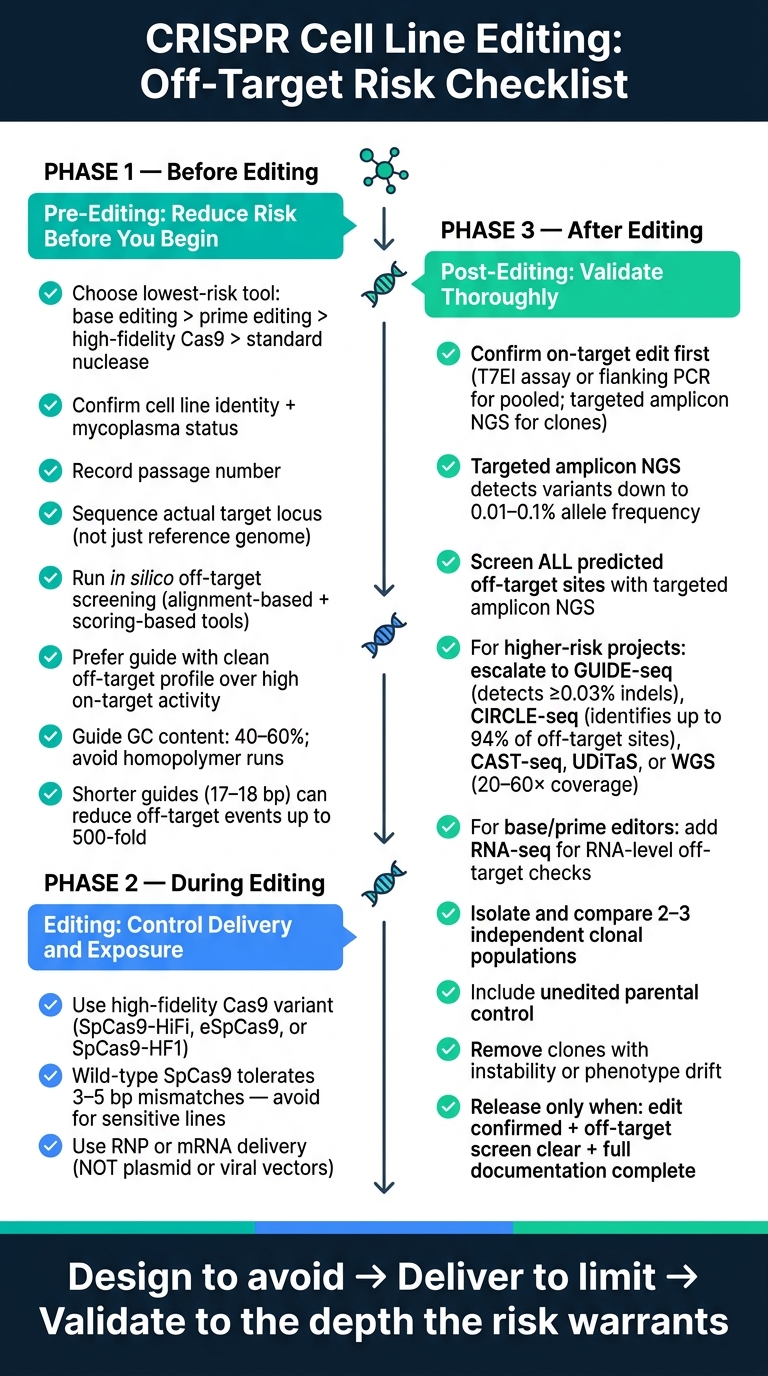
CRISPR脱靶风险控制:细胞系编辑的三阶段检查表
编辑前检查表:在编辑开始前降低风险
定义编辑目标并选择最低风险的编辑方法
在订购任何试剂之前,明确编辑的目的是什么。敲除、敲入、单核苷酸变化和转录调控不具有相同的脱靶风险。它们也不需要相同的工具。
广泛的风险顺序是简单明了的。形成双链断裂的核酸酶如Cas9和Cas12位于风险的高端,因为它们可能导致大规模的缺失、易位和DNA损伤反应[1] [7]. 碱基编辑器和主要编辑器使用nickases,因此它们避免了DSBs并降低了结构变异的风险 [1][5]. 对于转录调控,表观遗传编辑器如与转录修饰因子融合的dCas9保持DNA序列不变[1].
实际规则很简单:使用最少基因毒性的方法,同时仍能实现所需的编辑。对于单核苷酸变化,CBEs或ABEs比HDR更合适,因为HDR仍可能引入插入缺失[3][1]. 对于替换和小插入或缺失,主要编辑通常显示出比标准CRISPR-Cas9更低的脱靶活性 [1]. 如果必须使用核酸酶,请选择高保真变体,例如SpCas9-HiFi, eSpCas9, 或SpCas9-HF1 [1][6].
一旦编辑方法确定,锁定工作细胞系和确切的目标序列。
确认细胞系身份、历史和目标位点序列
如果细胞系被误认或交叉污染,其余的工作流程就会开始动摇。即使是设计良好的引导RNA也无法挽救不良的起始材料。在任何编辑开始之前,检查细胞系身份。同时,确认支原体状态并记录当前传代次数,因为高传代细胞可能会改变基因组稳定性和编辑效率[1][6].
同样重要的是,不要仅依赖参考基因组。对工作细胞系中的确切目标位点进行测序。此步骤有助于发现可能阻碍引导结合或创建新的脱靶位点的SNP或插入缺失[1][6].
之后,进入引导设计阶段。
在选择试剂之前,进行计算机模拟脱靶筛选
一旦确认目标位点,在进行湿实验之前,先在计算机上筛选候选引导RNA。使用基于比对的工具,如Cas-OFFinder或FlashFry, 以及基于评分的工具,如CFD评分或DeepCRISPR. 第一组工具有助于找到具有序列同源性的基因组位点。第二组工具有助于根据预测的切割概率对这些位点进行排名[1] [5].
当指南被列入候选名单时,较少的脱靶特征应优于原始的靶向效率。一个具有70%靶向效率且无预测脱靶的指南比一个具有90%效率且有多个高风险位点的指南更安全 [6]. 在某些情况下,将指南长度从20 bp缩短到17-18 bp可以在不损失太多靶向准确性的情况下减少多达500倍的脱靶事件 [5]. 目标是GC含量在40%到60%之间,并避免连续四个或更多相同碱基 [6][5].
话虽如此,计算机筛选是有限的。它不能很好地考虑染色质状态、细胞周期或细胞特定的背景 [1][6][4]. 将其视为一种过滤器,而不是证明。它缩小了范围,但并不能替代实验确认。
将最高风险的预测位点纳入编辑和验证计划。
sbb-itb-ffee270
编辑清单:控制编辑器选择、交付和暴露
使用高特异性编辑器和排名良好的引导RNA
从预测的脱靶候选名单开始,并用它来选择编辑器。在大多数情况下,高保真度的SpCas9变体 - SpCas9-HiFi、eSpCas9或SpCas9-HF1 - 比野生型SpCas9更好的默认选择 [6][1]. 野生型SpCas9可以容忍多达三到五个碱基对错配 , 特别是在PAM远端区域, 这在敏感细胞系中会产生显著的脱靶风险 [3].
这里有一个简单的规则:使用最不活跃的高保真编辑器,但仍能实现预期的编辑。
对于基础编辑器,分别跟踪旁观者编辑和RNA非目标效应,与DNA非目标风险 分开[1] [8] . 这些是不同的故障模式,需要单独检查。如果可以在不产生双链断裂 的情况下进行编辑,, 基础编辑或引导编辑可能更适合高风险工作流程[1][8].
一旦选择了编辑器,接下来的任务就是尽量缩短其在细胞内的时间。
通过瞬时传递和最小有效剂量限制编辑器的持久性
编辑器的持久性与编辑器的选择同样重要。编辑器在细胞中保持活跃的时间越长,它在低概率位点起作用的时间就越多。这使得交付格式成为一个主要的控制点。
使用瞬时交付,例如RNPs或mRNA, ,避免使用质粒DNA或病毒载体 ,因为它们会延长编辑器的表达时间 [1][5] . 在实践中,RNP交付应为默认选择 [6].
剂量也很重要。高核酸酶浓度增加了在低敏感性非靶位点切割的机会[5] . 使用最低有效剂量. 如果转染效率较差,不要只是增加试剂量并寄希望于最好的结果。那通常是转移问题而不是解决问题。
为高风险工作流程增加精确度保障措施
某些工作流程需要额外的保护措施。这对于靠近癌基因, 肿瘤抑制因子, 或在p53敏感细胞系, 中的目标尤其如此,因为一次脱靶事件可能会导致巨大的代价[1][6][3].
有用的保障措施包括:
- 配对切口酶, 需要两个相邻的切口。单个脱靶切口通常会在没有突变的情况下修复,因此与标准核酸酶设置相比,脱靶风险大大降低[4][1].
- 可诱导的、光控的或分裂的Cas9系统, 在传递效率高且暴露时间必须保持短暂时,有助于将编辑器活动保持在一个紧凑的窗口内[1].
- 抗CRISPR (Acr) 蛋白, 作为关闭开关。这些天然存在的Acr蛋白可以在定义的时间间隔后使CRISPR-Cas复合物失活,为编辑器活动提供分子制动[1].
编辑后检查清单:检测脱靶事件并验证克隆
使用靶向测序筛选预测的脱靶位点
编辑完成后,首先确认目标位点的预期变化。对于快速的初步筛查,可以在混合细胞中使用错配切割分析,例如T7内切酶I, 限制性消化或侧翼PCR。只需小心解释:每种方法都有灵敏度限制,尤其是对于罕见编辑或纯合变体[9].
对于克隆级验证,靶向扩增子NGS是标准。它为您提供等位基因频率的定量视图,并且可以检测到低至0.01%到0.1%的变体[3].
使用靶向扩增子NGS对每个预测的脱靶位点进行测序。这应该是默认的验证步骤。
当项目风险较高时,升级到全基因组或结构分析
逐个位点筛查并不总是足够的。如果编辑器、目标位点或细胞系暗示隐藏风险,请转向可以检测到您未提前预测的事件的分析。
全基因组发现分析如GUIDE-seq和CIRCLE-seq不需要事先的脱靶位点列表。 GUIDE-seq可以检测到频率低至0.03%的脱靶位点 [2]. CIRCLE-seq可以识别出多达94%的脱靶位点体外 [3] . 当细胞类型的背景可能掩盖脱靶活性时,这些方法很有用。
如果您担心大规模重排,标准扩增子读取可能会错过主要问题。缺失、倒位和易位需要为结构变化设计的检测方法,例如CAST-seq 和UDiTaS [1].
全基因组测序(WGS)是最广泛的选择。它可以检测基因组中的插入缺失、结构变异和拷贝数变化 [1] . 权衡在于深度和成本:通常需要20–60×覆盖率,这使其不适合常规筛查大规模人群[1].
使用靶向扩增子NGS用于预测位点。对于高风险项目,转向全基因组或结构分析。对于碱基或主要编辑器,添加RNA-seq以检查RNA水平的脱靶效应。
选择多个独立克隆并记录释放标准
在序列检查后,在多个克隆中测试表型。
不要仅依赖单个编辑克隆。分离并扩增至少两个到三个独立克隆群体 ,并与未编辑的亲本对照进行比较[4][9]. 去除显示不稳定性或表型漂移的克隆[3]. 然后使用靶向扩增子NGS确认所需等位基因状态的预期编辑,无论是杂合还是纯合 [9].
文档记录不是最后的管理工作。它是克隆发布的一部分。记录亲本背景, sgRNA设计、核酸酶变体、递送方法和所有QC结果 [3]. 只有在确认预期编辑、预测的脱靶位点清晰且完整记录到位时,克隆才能继续前进。
基因组编辑 CRISPR: 如何有效地最小化脱靶效应
结论:一个三阶段的清单用于更清洁的细胞系编辑
总而言之,该清单将脱靶控制视为一个分阶段的过程,而不是一次性的QC检查。目标很简单:早期降低风险,限制编辑器活动,然后验证结果。
验证深度应与风险相匹配。仅释放在预期等位基因状态下确认的多个独立克隆。
常见问题解答
为什么不依赖一个克隆?
依赖单个克隆是有风险的。CRISPR编辑并不是完全特异的, 因此可能引入意外的非目标突变。
这就是为什么团队通常会扩展多个克隆群体。这样做可以更容易找到携带预期目标编辑且没有有害非目标变化的细胞系。
还有另一个原因:细胞系可能表现出遗传异质性。对多个克隆进行测序有助于确认预期的纯合敲除或其他目标位点修改在目标基因座上存在。
什么时候扩增子NGS就足够了?
当您需要一种有针对性且具有成本效益的方法来确认计算工具或其他筛选方法标记的潜在非目标位点时,基于扩增子的下一代测序通常就足够了。
全基因组测序仍然是完全量化非目标效应的唯一方法。但对于许多应用来说,这种级别的分析并不是必需的。
我该如何选择最安全的编辑器?
选择仍能很好地切割您的目标位点的最低活性CRISPR核酸酶变体。
仅靠预测无法选择最佳变体。唯一可靠的方法是对选定的核酸酶变体进行小规模筛选,并通过下一代测序读取编辑结果。.
对于培养肉的研发,这为您提供了一个实际的前进路径:从一个简短的变体列表开始,然后逐步测试较弱的变体,直到找到仍能有效编辑目标位点的最低活性选项.









