

Hồ sơ lô hàng rất quan trọng đối với việc tuân thủ và an toàn sản phẩm. Chúng ghi lại từng bước của quá trình sản xuất, đảm bảo các tiêu chuẩn quy định được đáp ứng. Đối với các nhà sản xuất thịt nuôi cấy, duy trì vô trùng và hồ sơ chi tiết là điều không thể thương lượng. Kiểm tra của FDA thường nêu bật các vấn đề như thiếu dữ liệu, đánh giá không đầy đủ và hành động khắc phục kém, có thể dẫn đến cảnh báo hoặc gián đoạn.
Điểm chính cần lưu ý:
- Hồ sơ lô hàng: Hai loại - Hồ sơ Lô Hàng Chính (MBR) (công thức) và Hồ sơ Sản Xuất Lô Hàng (BPR) (thực hiện).
- Vấn đề thường gặp: Lỗi con người (50% vấn đề), thiếu kiểm tra trong quá trình, đánh giá không đầy đủ và hệ thống CAPA (Hành động Khắc phục và Phòng ngừa) kém.
- Tiêu chuẩn FDA: Tuân thủ các nguyên tắc ALCOA+ (Có thể quy kết, Dễ đọc, Đồng thời, Nguyên bản, Chính xác, Hoàn chỉnh, Nhất quán, Bền vững, Có sẵn) là bắt buộc.
- Giải pháp: Kiểm toán độc lập, hồ sơ lô điện tử và xác minh nhà cung cấp nghiêm ngặt có thể giảm thiểu lỗi và cải thiện tuân thủ.
Các công ty thịt nuôi cấy như UPSIDE Foods đã đặt ra một tiêu chuẩn bằng cách đảm bảo tài liệu chi tiết, truy xuất nguồn gốc đầu vào và các biện pháp khắc phục nhanh chóng. Bằng cách học hỏi từ những thực hành này, các nhà sản xuất có thể tránh được những cạm bẫy quy định và duy trì tiêu chuẩn chất lượng cao.
Hướng dẫn Toàn diện về Tài liệu và Lưu trữ Hồ sơ cho FDA Tuân thủ trong Khoa học Đời sống
sbb-itb-ffee270
Vấn đề Thường gặp trong Tài liệu Hồ sơ Lô
Các báo cáo kiểm tra của FDA liên tục nêu bật một vấn đề tái diễn: sự sai lệch trong việc xem xét hồ sơ sản xuất nằm trong số các thiếu sót GMP hàng đầu được các cơ quan quản lý trích dẫn [7]. Đối với các nhà sản xuất thịt nuôi cấy, những thiếu sót này vượt xa các lỗi hành chính đơn thuần - chúng đe dọa khả năng chứng minh điều kiện vô trùng bền vững. Những vấn đề này xuất hiện dưới nhiều hình thức khác nhau, như được minh họa trong các ví dụ dưới đây.
Đánh Giá Không Hoàn Chỉnh và Không Tuân Thủ
Một vấn đề phổ biến là sự thất bại của các Đơn Vị Kiểm Soát Chất Lượng trong việc xem xét kỹ lưỡng hồ sơ lô hàng. Thay vì là một phần không thể thiếu của quy trình phát hành, các đánh giá thường xảy ra một cách phản ứng - chỉ sau khi một vấn đề sản phẩm đã xuất hiện [7]. Cách tiếp cận này để lại những khoảng trống đáng kể trong hồ sơ sản xuất.
Ví dụ, Davis City Pharmacy đã nhận được một quan sát 483 của FDA do hồ sơ lô hàng thiếu các chi tiết quan trọng như số lượng thành phần, các bước vận hành và chữ ký của nhân viên. Tương tự, CAPS đã bị chỉ trích vì thiếu chữ ký cần thiết và xác minh của người đánh giá trong các mục quan trọng [3]. Những sai sót này không phải là những sự cố đơn lẻ; các nghiên cứu cho thấy rằng khoảng 52% vi phạm tài liệu leo thang khi thiếu hệ thống quản lý quy trình sinh học mạnh mẽ [3].
"Không phải sự phức tạp của quy trình gây ra các trích dẫn - mà là sự không nhất quán, không đầy đủ và giám sát kém." - Dịch vụ Tư vấn & Kiểm toán GXP [5]
Thiếu Hồ Sơ Kiểm Tra Trong Quá Trình
Một thiếu sót thường xuyên khác là thiếu tài liệu đúng đắn cho các kiểm tra trong quá trình. Những hồ sơ này rất quan trọng, đặc biệt là tại các điểm kiểm soát quan trọng trong các hoạt động vô trùng. Ví dụ, Trung tâm Pha chế Vô trùng Nephron đã bị trích dẫn vì không ghi lại các bước mặc đồ cần thiết và các quy trình vô trùng trong hồ sơ lô của họ [3]. Đối với các nhà sản xuất thịt nuôi cấy, nơi mà sự vô trùng là tối quan trọng, những thiếu sót như vậy khiến không thể xác nhận tuân thủ các biện pháp kiểm soát ô nhiễm.
Amphastar cũng bị cảnh báo vì không điều tra hoặc ghi chép các biến động sản lượng bất ngờ hoặc sự khác biệt đầu ra [3]. Rủi ro của những sai sót như vậy là rõ ràng. Trong một trường hợp, một cơ sở dược phẩm không xác định vào năm 2024/2025 đã bị phát hiện lưu trữ hồ sơ lô hoàn chỉnh trên kệ mở và bàn làm việc. Các điều tra viên của FDA phát hiện các trang bị thiếu, bao gồm bảy trang từ một hồ sơ duy nhất, và toàn bộ phần "Dung dịch Tổng hợp" bị thiếu từ một hồ sơ khác [6].
Thất bại trong CAPA và Xác minh GMP của Nhà cung cấp
Vượt ra ngoài các lỗi tài liệu, sự thiếu vắng các quy trình khắc phục hiệu quả và xác minh nhà cung cấp càng làm suy yếu độ tin cậy của hồ sơ lô.Khi xảy ra sai lệch trong sản xuất mà không có báo cáo Hành động Khắc phục và Phòng ngừa (CAPA) tương ứng, tính toàn vẹn của hồ sơ lô hàng bị ảnh hưởng [7]. Ví dụ, Eugia Pharma Specialities Limited, được kiểm tra từ ngày 22 tháng 1 đến ngày 2 tháng 2 năm 2024, đã nhận được FDA 483 vì không xem xét đầy đủ các sai lệch. Hệ thống CAPA không hiệu quả và các cuộc điều tra không hoàn chỉnh của họ đã dẫn đến các vấn đề sản xuất lặp đi lặp lại, buộc phải cải tổ hoàn toàn các quy trình điều tra và CAPA của họ [9].
Tương tự, trong một cuộc kiểm tra từ ngày 26 tháng 9 đến ngày 25 tháng 10 năm 2023, Stokes Healthcare Inc. đã thể hiện quản lý sai lệch kém. Công ty đã không mở rộng điều tra đến tất cả các lô hàng bị ảnh hưởng và trì hoãn hoàn thành các phân tích của họ [9].
"Một sai lệch mà không có báo cáo CAPA hoặc báo cáo sai lệch tương ứng? Đó là một thất bại trong tuân thủ." - GXP Kiểm toán & Dịch vụ Tư vấn [5]
Vấn đề xác minh nhà cung cấp thêm một lớp phức tạp khác. Empower Clinic Services LLC đã bị trích dẫn trong một cuộc kiểm tra từ ngày 18 tháng 7 đến ngày 5 tháng 8 năm 2022 vì các quy trình kiểm soát chất lượng không đầy đủ, bao gồm cả việc không đủ tiêu chuẩn nhà cung cấp và quy trình điều tra kém [9]. Đối với các nhà sản xuất thịt nuôi cấy, những người phụ thuộc vào môi trường tăng trưởng, dòng tế bào và các đầu vào quan trọng khác, đảm bảo tuân thủ GMP của nhà cung cấp là rất quan trọng để duy trì tính toàn vẹn của hồ sơ lô.
Yêu cầu của FDA đối với Hồ sơ Lô
Quy tắc của FDA đối với hồ sơ lô xoay quanh 21 CFR Phần 117, đặt ra tiêu chuẩn cơ bản cho an toàn thực phẩm.Khi nói đến thịt nuôi cấy, nơi mà việc duy trì vô trùng trong giai đoạn nuôi cấy tế bào là rất quan trọng, tài liệu thường cần phải đáp ứng các tiêu chuẩn nghiêm ngặt hơn của Phần 111 hoặc Phần 211, ngoài Phần 117 [10][14]. Điều này nhấn mạnh tầm quan trọng của việc tài liệu chính xác để đảm bảo an toàn và hiệu quả của sản xuất thịt nuôi cấy.
Tiêu Chuẩn Cốt Lõi cho Hồ Sơ Lô Hàng
Mỗi lô hàng yêu cầu hai tài liệu chính:
- Hồ Sơ Lô Hàng Chính (MBR): Mẫu được phê duyệt mô tả quy trình sản xuất.
- Hồ Sơ Sản Xuất Lô Hàng (BPR): Một hồ sơ chi tiết về những gì thực sự xảy ra trong quá trình sản xuất [12][2].
BPR phải bao gồm các chi tiết cụ thể như số lô hoặc số mẻ, chi tiết thiết bị, ngày làm sạch, mã định danh thành phần, các phép đo chính xác và so sánh giữa sản lượng thực tế và lý thuyết [10][14].
"Hồ sơ sản xuất mẻ phải tuân theo chính xác hồ sơ sản xuất chính và bạn phải thực hiện từng bước trong quá trình sản xuất mẻ." – 21 CFR 111.255 [12]
Mỗi bước quan trọng phải được ghi lại ngay lập tức, với chữ ký tắt của cả người thực hiện và người kiểm tra [10][11]. FDA yêu cầu tuân thủ các nguyên tắc ALCOA(+), nghĩa là hồ sơ phải Có thể quy trách nhiệm, Dễ đọc, Đồng thời, Nguyên bản và Chính xác - cũng như Hoàn chỉnh, Nhất quán, Bền vững và Có sẵn [1].
Nếu có bất kỳ sai lệch nào so với Hồ sơ Sản xuất Chính, nó phải được điều tra kỹ lưỡng. Điều này bao gồm việc ghi lại vấn đề, tiến hành phân tích nguyên nhân gốc rễ và thực hiện kế hoạch Hành động Khắc phục và Phòng ngừa (CAPA) [8] [1]. Đánh giá ban đầu về các sai lệch nên được ghi lại trong vòng 24–48 giờ sau khi phát hiện [8]. Đối với các cơ sở sử dụng hệ thống điện tử, tuân thủ 21 CFR Phần 11 là bắt buộc. Điều này bao gồm chữ ký điện tử đã được xác nhận và các dấu vết kiểm toán an toàn, có dấu thời gian [8] [1].
Quy trình Lưu trữ và Xem xét Hồ sơ
Quy trình lưu trữ và xem xét hồ sơ đúng cách là rất quan trọng để duy trì tuân thủ và đảm bảo an toàn sản phẩm.Trong sản xuất vô trùng, như sản xuất thịt nuôi cấy, mọi chi tiết trong hồ sơ lô hàng phải trải qua quá trình xem xét tỉ mỉ. Đội ngũ Kiểm soát Chất lượng (QC) chịu trách nhiệm xem xét tất cả hồ sơ lô hàng, giám sát kết quả và dữ liệu thử nghiệm trước khi một lô hàng có thể được phê duyệt để phân phối [10] [13].
"Tất cả hồ sơ sản xuất và kiểm soát sản phẩm thuốc phải được đơn vị kiểm soát chất lượng xem xét và phê duyệt trước khi một lô hàng được phát hành hoặc phân phối." – 21 CFR 211.192 [2]
Các nhà sản xuất thường đặt mục tiêu hoàn thành 95% việc xem xét lô hàng trong vòng 30 ngày kể từ khi sản xuất [2] . Tuy nhiên, đối với các quy trình vô trùng phức tạp hơn liên quan đến thịt nuôi cấy, việc xem xét thường mất 7–10 ngày, với các cơ sở hoạt động hiệu quả cao đạt được thời gian dưới 7 ngày [2]. Hệ thống hồ sơ lô điện tử có thể tăng tốc đáng kể các đánh giá này, chẳng hạn như những hệ thống được tích hợp vào hệ thống sản xuất thịt nuôi cấy, - giảm thời gian xuống một nửa so với phương pháp dựa trên giấy - miễn là chúng được xác nhận để đáp ứng các yêu cầu của Phần 11 và duy trì tính toàn vẹn của dữ liệu[1].
Những điều các công ty thịt nuôi cấy được FDA phê duyệt đã làm đúng
Các công ty thịt nuôi cấy được FDA phê duyệt đã đặt ra tiêu chuẩn cao bằng cách áp dụng các thực hành giải quyết thách thức về tài liệu và đáp ứng các tiêu chuẩn an toàn nghiêm ngặt.
Khi UPSIDE Foods trở thành công ty thịt nuôi cấy đầu tiên vượt qua cuộc tham vấn trước thị trường của FDA vào tháng 11 năm 2022, họ đã thiết lập một mô hình cho ngành công nghiệp.FDA đã phát hành một lá thư "không có câu hỏi nào thêm" sau khi xem xét kỹ lưỡng quy trình sản xuất của họ, bao gồm việc thiết lập dòng tế bào, ngân hàng tế bào, kiểm soát sản xuất, và tất cả các thành phần và đầu vào [16]. Thành tựu này nhấn mạnh tầm quan trọng của việc tài liệu chi tiết để đáp ứng các yêu cầu nghiêm ngặt của FDA.
Đáp ứng Tiêu chuẩn Vô trùng và Tuân thủ
Thành tựu nổi bật của UPSIDE Foods là cách tiếp cận toàn diện của họ đối với khả năng truy xuất nguồn gốc đầu vào. Mỗi thành phần sản xuất đều được ghi chép cẩn thận, đảm bảo một chuỗi trách nhiệm rõ ràng từ dòng tế bào ban đầu đến sản phẩm cuối cùng [16]. Mức độ minh bạch này cho phép các nhà đánh giá của FDA theo dõi từng bước của quy trình sản xuất, xác nhận rằng tất cả các tiêu chuẩn an toàn đều được đáp ứng một cách nhất quán.
"Cuộc tham vấn trước thị trường của FDA với công ty bao gồm việc đánh giá quy trình sản xuất của công ty và vật liệu tế bào nuôi cấy được tạo ra bởi quy trình sản xuất, bao gồm việc thiết lập dòng tế bào sơ cấp và bất tử và ngân hàng tế bào, kiểm soát sản xuất, và tất cả các thành phần và đầu vào." – U.S. Cục Quản lý Thực phẩm và Dược phẩm [16]
Các công ty thành công khác đã làm theo bằng cách thực hiện tài liệu quy trình vô trùng chi tiết. Điều này bao gồm các bước quan trọng như quy trình mặc đồ bảo hộ và thao tác vô trùng [3]. Không giống như những thất bại tài liệu trước đó, các công ty này đã áp dụng hệ thống đánh giá phân tầng, bao gồm kiểm tra của người vận hành, giám sát sản xuất và đánh giá của đơn vị chất lượng, để phát hiện lỗi tiềm ẩn trước khi phát hành lô hàng [15]. Hệ thống hồ sơ lô điện tử cũng đóng vai trò then chốt, thực thi các xác nhận bắt buộc ở mỗi giai đoạn và duy trì các dấu vết kiểm toán không thể thay đổi theo yêu cầu của 21 CFR Phần 11 [3][2].
Những thực hành nghiêm ngặt này tự nhiên mở rộng vào cách các công ty xử lý các sai lệch và thất bại.
Quy trình CAPA cho các thất bại của lô hàng
Khi các lô hàng không đạt tiêu chuẩn, các công ty được FDA phê duyệt đã hành động nhanh chóng và có hệ thống. Quy trình Hành động Khắc phục và Phòng ngừa (CAPA) của họ bao gồm phân tích nguyên nhân gốc rễ chính thức, đánh giá tác động và các hành động khắc phục được ghi chép rõ ràng [3]. Bất kỳ sai lệch nào cũng được quản lý trong một khung đảm bảo chất lượng tích hợp, đảm bảo rằng tất cả các vấn đề đều được điều tra kỹ lưỡng, biện minh và ghi chép trước khi sản xuất tiếp tục [2].
Nhìn về phía trước, tính toàn vẹn dữ liệu sẽ trở thành trọng tâm chính của các hành động thực thi của FDA cho năm 2024–2025 [1].
Cách Cải Thiện Thực Hành Hồ Sơ Lô Hàng của Bạn
Tăng cường thực hành hồ sơ lô hàng đòi hỏi tài liệu chính xác để giải quyết các thất bại thường gặp được xác định trong các cuộc kiểm tra của FDA. Dưới đây là một số chiến lược để giải quyết các thách thức chính.
Thực Hiện Kiểm Toán Hồ Sơ Lô Hàng Độc Lập
Kiểm toán bên thứ ba thường xuyên có thể phát hiện các vấn đề mà các đánh giá nội bộ có thể bỏ qua. Bắt đầu bằng cách tập trung vào các hệ thống quan trọng như Hệ Thống Quản Lý Thông Tin Phòng Thí Nghiệm (LIMS), Hệ Thống Thực Thi Sản Xuất (MES), và Hoạch Định Nguồn Lực Doanh Nghiệp (ERP). Ưu tiên các tài liệu có tác động quy định cao, chẳng hạn như hồ sơ kiểm tra phát hành, dữ liệu ổn định, và hồ sơ sản xuất lô hàng.
Một phương pháp hiệu quả là kiểm tra thu hồi mẫu.Chọn ngẫu nhiên các lô hàng gần đây và tái cấu trúc lịch sử sản xuất và phòng thí nghiệm của chúng. Điều này có thể giúp xác định dữ liệu bị thiếu, chữ ký không đầy đủ hoặc các khoảng trống trong tài liệu có thể dẫn đến các trích dẫn quy định. Đối chiếu các dấu vết kiểm toán do hệ thống tạo ra với các mục nhập thủ công để xác định các thay đổi hoặc xóa không được phép.
Xem xét tất cả các báo cáo Ngoài Tiêu Chuẩn (OOS) và Ngoài Xu Hướng (OOT) từ năm qua. Đánh giá xem các phân tích nguyên nhân gốc rễ có kỹ lưỡng hay không và nếu các Hành Động Khắc Phục và Phòng Ngừa (CAPA) đã được thực hiện đầy đủ. Đáng chú ý rằng các vấn đề về tài liệu chiếm 21% các thư cảnh báo của FDA, trong khi lỗi con người đóng góp 50% các vấn đề về hồ sơ lô hàng trong sản xuất dược phẩm [2].
"Không phải sự phức tạp của quy trình gây ra các trích dẫn - mà là sự không nhất quán, không đầy đủ và giám sát kém." – Dịch vụ Tư vấn Kiểm toán GXP & [5]
Mô phỏng các cuộc kiểm tra quy định thông qua các đánh giá giả định định kỳ. Thực hành này giúp các nhóm nhận ra sự không nhất quán và các vấn đề tiềm ẩn về tính toàn vẹn dữ liệu trước khi có một cuộc kiểm toán thực sự. Đảm bảo tất cả hồ sơ tuân theo các nguyên tắc ALCOA+: Có thể quy kết, Dễ đọc, Đồng thời, Nguyên bản, Chính xác, Hoàn chỉnh, Nhất quán, Bền vững và Có sẵn.
Một khi tính toàn vẹn của tài liệu đã vững chắc, hãy tập trung vào việc xác minh chất lượng của tất cả các đầu vào sản xuất.
Kiểm tra Tất cả Đầu vào về Ô nhiễm Vi sinh
Kiểm tra độc lập về độ vô trùng và hiệu lực cho tất cả các đầu vào là cần thiết - không chỉ dựa vào Chứng chỉ Phân tích (CoAs) của nhà cung cấp. Điều này đặc biệt quan trọng đối với các nhà sản xuất thịt nuôi cấy, vì ô nhiễm có thể gây nguy hiểm cho toàn bộ lô hàng.
Ví dụ, vào tháng 2 năm 2013, Central Admixture Pharmacy Services đã bị FDA cảnh cáo do kiểm soát vi sinh không đầy đủ trong quá trình phát hành lô sản phẩm vô trùng. Công ty đã phải đưa ra các quy trình kiểm soát vi sinh chi tiết trong Quy trình Hoạt động Tiêu chuẩn (SOPs) của mình [4].
Các điểm kiểm tra vi sinh trong quá trình có thể ngăn chặn sự phụ thuộc quá mức vào việc kiểm tra sản phẩm cuối cùng. Tích hợp các điểm kiểm tra này vào SOPs phát hành lô và duy trì tài liệu đồng thời nghiêm ngặt. Ghi lại tất cả kết quả kiểm tra và các bước sản xuất khi chúng xảy ra để tránh việc ghi lùi ngày hoặc nhập liệu chậm trễ, điều này có thể dẫn đến cảnh cáo từ FDA.
Giữ hồ sơ nhà cung cấp toàn diện, bao gồm CoAs, báo cáo kiểm toán, thỏa thuận chất lượng và lịch sử của bất kỳ sai lệch nào liên quan đến nguyên liệu đầu vào.
Tăng cường thực hành ghi chép lô sản phẩm hơn nữa bằng cách điều chỉnh các quy trình với các tiêu chuẩn đã được thiết lập như HACCP và GCCP.
Đồng bộ Hồ sơ với Tiêu chuẩn HACCP và GCCP
Việc tích hợp các nguyên tắc của Điểm Kiểm Soát Quan Trọng Phân Tích Nguy Cơ (HACCP) vào hồ sơ lô hàng đảm bảo các biến số quan trọng của quy trình được giám sát và ghi lại trong suốt quá trình sản xuất. Điều này bao gồm việc thiết lập các điểm kiểm tra vi sinh trong quá trình thay vì chỉ dựa vào các thử nghiệm giai đoạn cuối.
Đối với các nhà sản xuất thịt nuôi cấy, tuân thủ các tiêu chuẩn Thực Hành Nuôi Cấy Tế Bào Tốt (GCCP) cũng quan trọng không kém. Hồ sơ lô hàng nên bao gồm chi tiết về các thao tác vô trùng, quy trình mặc đồ bảo hộ và giám sát môi trường liên quan đến tiêu chí phát hành lô hàng [3][4]. Các bước này giúp duy trì sự tuân thủ và đảm bảo an toàn sản phẩm.
Dữ liệu ngành cho thấy rằng 52% vi phạm tài liệu gia tăng khi không có phần mềm sản xuất lô hàng phù hợp [3][4]. Một ví dụ điển hình: vào tháng 2 năm 2023, Trung tâm Pha chế Vô trùng Nephron đã nhận được một quan sát từ FDA do thiếu các quy trình kiểm soát để xác minh các biến số quy trình quan trọng trước khi phát hành lô hàng [4]. Điều này nhấn mạnh sự cần thiết của việc tài liệu chủ động phù hợp với các tiêu chuẩn được công nhận.
Chuyển đổi sang Hồ sơ Lô Điện tử (EBR) có thể giảm đáng kể lỗi tài liệu - lên đến 50% - thông qua việc thu thập dữ liệu theo thời gian thực và quy trình tự động [2]. Các hệ thống này đánh dấu các kết quả kiểm tra vi sinh vật bị thiếu hoặc các đánh giá chưa hoàn chỉnh trước khi một lô hàng tiến hành, giảm thiểu lỗi do con người.
"FDA mong đợi các hồ sơ phải là ALCOA(+): Có thể quy trách nhiệm, Dễ đọc, Đồng thời, Nguyên bản, Chính xác - cộng với Hoàn chỉnh, Nhất quán, Bền vững và Có sẵn." – Atlas Compliance [1]
Mọi sự khác biệt hoặc sai lệch không giải thích được trong hồ sơ lô hàng nên được liên kết với một hệ thống điều tra chính thức và CAPA. Hạn chế quyền viết và xóa để bảo vệ tính toàn vẹn của dữ liệu kiểm tra vi sinh điện tử. Các nhà sản xuất cạnh tranh đặt mục tiêu xem xét và phát hành 95% lô hàng trong vòng 30 ngày kể từ khi hoàn thành sản xuất [2].
Những hành động này không chỉ giảm nguy cơ bị trích dẫn mà còn phù hợp với các tiêu chuẩn tài liệu nghiêm ngặt được nhấn mạnh trong các cuộc kiểm tra gần đây của FDA.
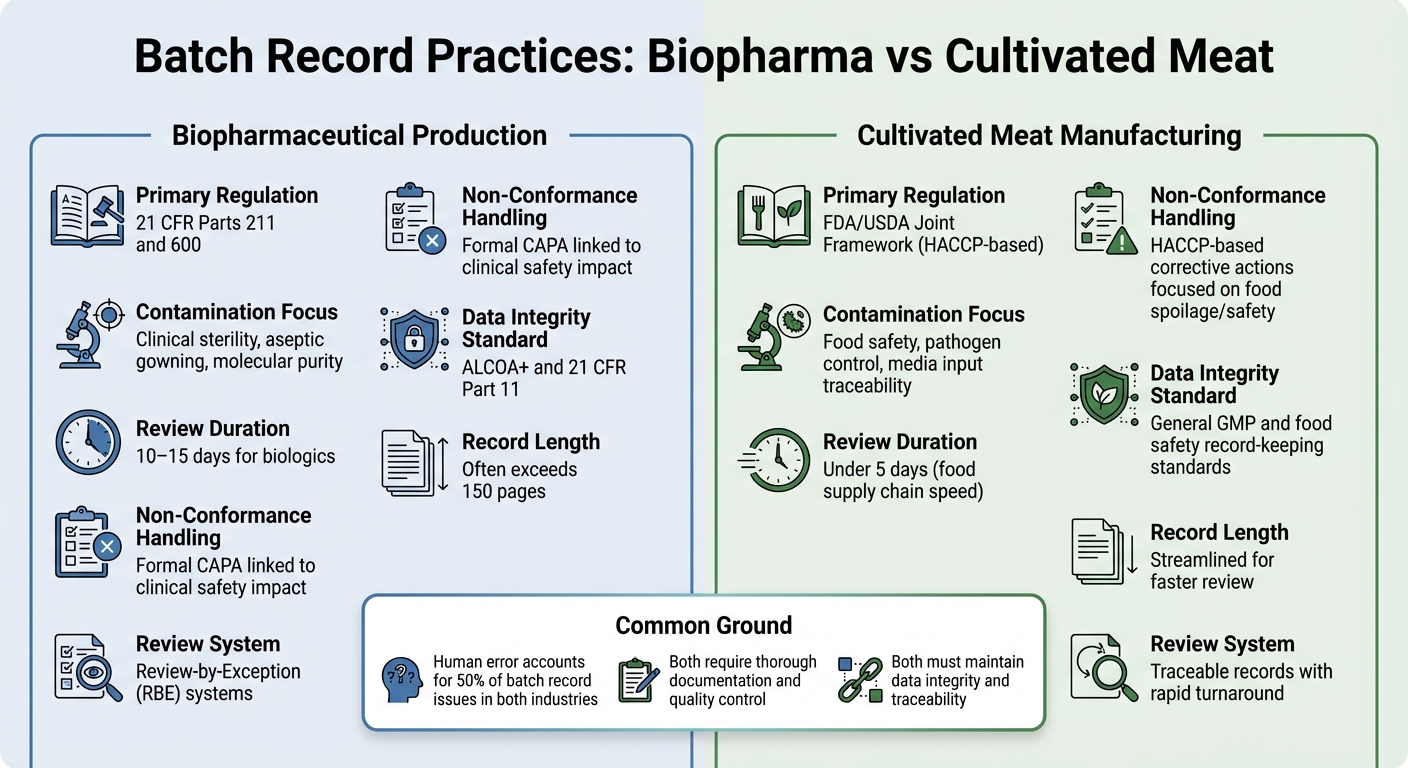
Biopharma vs Thịt nuôi cấy: Sự khác biệt trong hồ sơ lô hàng
So sánh yêu cầu hồ sơ lô hàng Biopharmaceutical và Thịt nuôi cấy
Xem xét sự khác biệt trong thực hành hồ sơ lô hàng giữa sản xuất dược phẩm sinh học và sản xuất thịt nuôi cấy cung cấp một cái nhìn rõ ràng hơn về cách yêu cầu quy định định hình ưu tiên tài liệu trong các ngành này.
Cả hai lĩnh vực đều yêu cầu tài liệu kỹ lưỡng, nhưng khung quy định và mục tiêu kiểm soát của họ khác nhau đáng kể. Trong dược phẩm sinh học, hồ sơ lô hàng được quy định chặt chẽ theo 21 CFR Phần 211 và 600, yêu cầu đơn vị Kiểm soát Chất lượng phải xem xét và phê duyệt tất cả các hồ sơ sản xuất và kiểm soát trước khi một lô hàng có thể được phát hành [2]. Các nhà sản xuất thịt nuôi cấy, mặt khác, thường tuân theo tiêu chuẩn HACCP và GCCP.Những điều này tập trung nhiều hơn vào an toàn thực phẩm và kiểm soát mầm bệnh, thay vì độ vô trùng cấp độ lâm sàng yêu cầu cho các sinh phẩm tiêm.
Hồ sơ lô hàng của ngành dược sinh học thường rất chi tiết, đôi khi vượt quá 150 trang, và quá trình xem xét có thể mất 10–15 ngày. Để đơn giản hóa điều này, nhiều công ty dược sinh học sử dụng hệ thống Review-by-Exception (RBE), hệ thống này tóm tắt các sai lệch chính thành một báo cáo ngắn gọn. Trong khi đó, các nhà sản xuất thịt nuôi cấy hướng tới hồ sơ có thể truy xuất nguồn gốc và có thể được xem xét trong vòng chưa đầy năm ngày, phản ánh tốc độ nhanh hơn của chuỗi cung ứng thực phẩm [2].
Nội dung của những hồ sơ này cũng làm nổi bật các ưu tiên khác nhau. Kiểm tra dược sinh học thường tập trung vào chi tiết xử lý vô trùng, chẳng hạn như quy trình mặc đồ bảo hộ và kiểm soát môi trường. Ngược lại, hồ sơ thịt nuôi cấy phải nhấn mạnh đầu vào môi trường và kiểm tra vi sinh để đảm bảo an toàn thực phẩm.Đối với thịt nuôi cấy, thách thức nằm ở việc theo dõi các đầu vào môi trường phức tạp, ghi chép các xét nghiệm vi sinh cho tất cả các vật liệu, và đáp ứng các giới hạn an toàn thực phẩm quan trọng - mà không cần tuân thủ các yêu cầu vô trùng nghiêm ngặt của dược phẩm.
Xu hướng ô nhiễm và không phù hợp
| Tính năng | Sản xuất dược phẩm sinh học | Sản xuất thịt nuôi cấy |
|---|---|---|
| Quy định chính | 21 CFR Phần 211 và 600[2] | Khuôn khổ chung FDA/USDA (dựa trên HACCP) |
| Tập trung vào ô nhiễm | Vô trùng lâm sàng, mặc áo vô trùng, độ tinh khiết phân tử[2] | An toàn thực phẩm, kiểm soát mầm bệnh, truy xuất nguồn gốc đầu vào môi trường |
| Thời gian xem xét | 10–15 ngày đối với sinh phẩm[2] | Dưới 5 ngày (tốc độ chuỗi cung ứng thực phẩm) |
| Xử lý không phù hợp | CAPA chính thức liên quan đến tác động an toàn lâm sàng [2] | Hành động khắc phục dựa trên HACCP tập trung vào hư hỏng/an toàn thực phẩm |
| Tiêu chuẩn Tính toàn vẹn Dữ liệu | ALCOA+ và 21 CFR Phần 11 [1] | Tiêu chuẩn ghi chép GMP chung và an toàn thực phẩm |
Mặc dù tỷ lệ lỗi của con người tương tự trong cả hai ngành - khoảng 50% vấn đề hồ sơ lô xuất phát từ sai lầm của con người [2] - nhưng mức độ quan trọng là khác nhau.Trong ngành dược sinh học, ngay cả một sai lệch không được ghi chép cũng có thể gây ra những hậu quả nghiêm trọng đối với an toàn của bệnh nhân. Đối với thịt nuôi cấy, rủi ro nhiễm bẩn chủ yếu liên quan đến các mầm bệnh từ thực phẩm và sự hư hỏng, có thể ảnh hưởng đến toàn bộ quy trình sản xuất.
Kết luận
Hồ sơ lô hàng đóng vai trò là nhật ký chính thức cho mỗi quy trình sản xuất thịt nuôi cấy - nếu một bước không được ghi lại, các cơ quan quản lý sẽ coi như chưa thực hiện [6][3]. Điều này nhấn mạnh tầm quan trọng của việc ghi chép chính xác và kiểm soát chất lượng nghiêm ngặt.
Các cuộc kiểm tra của FDA nhấn mạnh rằng tính toàn vẹn của dữ liệu phải phù hợp với các nguyên tắc ALCOA+ [1]. Các đội Kiểm soát Chất lượng được yêu cầu xem xét và phê duyệt tất cả các hồ sơ sản xuất trước khi một lô hàng có thể được phát hành [2][17], và bất kỳ sai lệch nào cũng phải được điều tra kịp thời với một phân tích nguyên nhân gốc rễ được ghi lại [2][5]. Mặc dù lỗi của con người chiếm 50% các vấn đề về hồ sơ lô hàng, các đánh giá hai cấp độ và quy trình CAPA (Hành động Khắc phục và Phòng ngừa) có cấu trúc có thể giúp giảm thiểu những rủi ro này [2][5].
"Không phải sự phức tạp của quy trình gây ra các trích dẫn - mà là sự không nhất quán, không đầy đủ và giám sát kém." - Dịch vụ Tư vấn Kiểm toán GXP & [5]
Để vượt qua những thách thức này, các nhà sản xuất thịt nuôi cấy nên tập trung vào các cuộc kiểm toán độc lập, kiểm tra nghiêm ngặt các thành phần an toàn thực phẩm để phát hiện ô nhiễm vi sinh, và đảm bảo tài liệu tuân thủ các tiêu chuẩn HACCP và GCCP. Việc triển khai hệ thống ghi chép lô điện tử, được xác nhận theo 21 CFR Phần 11 [1], có thể giảm thiểu đáng kể lỗi và tăng tốc quá trình xem xét.
Môi trường pháp lý đòi hỏi sự chính xác, nhưng có thể điều hướng được.Bằng cách học hỏi từ những sai lầm của ngành dược phẩm sinh học - chẳng hạn như thiếu chữ ký tại Qinhuangdao Zizhu Pharmaceutical [17], thiếu xác minh kép tại Terumo Corp [18] , và tài liệu sai lệch không đầy đủ tại Torrent Pharmaceuticals [18] - các công ty sản xuất thịt nuôi cấy có thể thiết lập các hệ thống tuân thủ ngay từ đầu. Việc tích hợp những bài học này cho phép tuân thủ chủ động và chất lượng nhất quán. Lưu trữ hồ sơ an toàn, báo cáo sai lệch kịp thời và thực hiện các cuộc kiểm tra giả định thực tế sẽ đảm bảo hồ sơ lô hàng luôn sẵn sàng cho kiểm tra và các đợt sản xuất có thể truy xuất đầy đủ.
Để có thêm tài nguyên và hướng dẫn chuyên gia về duy trì tiêu chuẩn sản xuất cao trong sản xuất thịt nuôi cấy, hãy truy cập
Câu hỏi thường gặp
Một hồ sơ lô hàng cho thịt nuôi cấy nên bao gồm những gì?
Một hồ sơ lô hàng cho thịt nuôi cấy đóng vai trò như một nhật ký toàn diện của toàn bộ quy trình sản xuất. Nó phải bao gồm hướng dẫn xử lý chi tiết, ghi chép thực hiện từng bước, và ghi chú bất kỳ sai lệch nào xảy ra trong quá trình sản xuất. Ngoài ra, nó nên ghi lại kiểm tra trong quá trình và kiểm tra phát hành để xác nhận sản phẩm đáp ứng các tiêu chuẩn an toàn, chất lượng và quy định.
Làm thế nào chúng ta có thể chứng minh sự vô trùng bằng hồ sơ lô hàng?
Chứng minh sự vô trùng thông qua hồ sơ lô hàng liên quan đến việc kiểm tra kỹ lưỡng các quy trình tiệt trùng đã được ghi chép, kết quả kiểm tra và báo cáo kiểm soát chất lượng môi trường để đảm bảo chúng đáp ứng các yêu cầu quy định.Điều quan trọng là phải giải quyết bất kỳ sai lệch hoặc thử nghiệm thất bại nào thông qua các cuộc điều tra chi tiết và CAPAs (Hành động Khắc phục và Phòng ngừa). Quy trình này đảm bảo rằng mọi bước đã được tuân thủ và bất kỳ vấn đề nào đã được giải quyết đúng cách để duy trì tiêu chuẩn vô trùng.
Khi nào cần hồ sơ lô điện tử (Phần 11)?
Hồ sơ lô điện tử là cần thiết theo Phần 11 khi các hệ thống điện tử được sử dụng để ghi chép, điều tra và biện minh cho các sai lệch hồ sơ lô. Chúng đóng vai trò quan trọng trong việc đảm bảo tuân thủ 21 CFR Part 211.192 , bảo vệ tính toàn vẹn của dữ liệu, đáp ứng thời hạn điều tra và đảm bảo giám sát quản lý hiệu quả.









