

If you edit first and check later, you can fix an off-target change into the clone. I’d keep the workflow simple: choose the lowest-risk editing method, keep editor exposure short, and then test both predicted off-target sites and clone stability before release.
For bioprocess engineers, cell culture scientists, and cultivated meat R&D teams, the main point is straightforward. CRISPR systems can still cut at near-match sites, often with 3–6 mismatches tolerated, and those errors can carry into expanded single-cell clones. The article breaks risk control into three phases: before editing, during editing, and after editing.
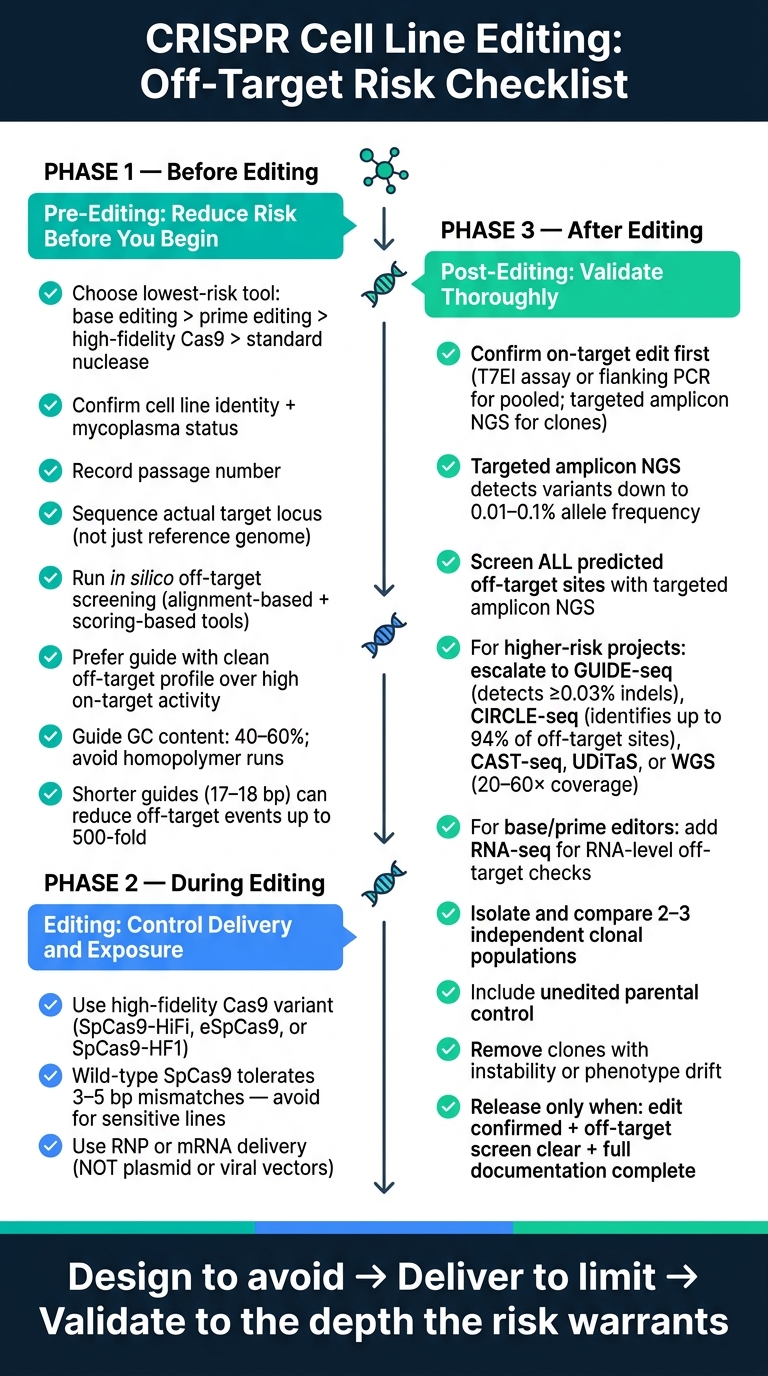
Here’s the full checklist in plain terms:
-
Pick the lowest-risk editing tool for the job
- Use base editing or prime editing when they can deliver the edit without a double-strand break
- Use dCas9-based modulation if you only need gene regulation
- If you need a nuclease, start with a high-fidelity Cas9 variant
-
Lock down the starting material
- Confirm cell line identity
- Check mycoplasma
- Record passage number
- Sequence the actual target locus in the working line, not just the reference genome
-
Screen guides before wet work
- Use alignment-based and scoring-based off-target tools together
- Prefer a guide with a cleaner off-target profile over one with only higher on-target activity
- Watch guide length, 40–60% GC content, and homopolymer runs
-
Limit exposure inside the cell
- Use RNP or mRNA delivery instead of plasmid or viral systems where possible
- Use the minimum effective dose
- Avoid extending editor persistence just to force transfection results
-
Add extra controls for higher-risk cases
- Consider paired nickases
- Use inducible, split-Cas9, or light-controlled systems when timing matters
- Add anti-CRISPR proteins as a shut-off step where needed
-
Validate properly after editing
- Confirm the on-target edit first
- Check every predicted off-target site with targeted amplicon NGS
- Move to GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, or WGS when project risk is higher
- For base or prime editors, add RNA-level checks where relevant
-
Do not release a single clone on sequence alone
- Compare 2–3 independent clones
- Use an unedited parental control
- Remove clones with instability or phenotype drift
- Release only when edit state, off-target screen, and records are all complete
A short way to think about it: design to avoid off-target cuts, deliver to limit time in-cell, then validate at the depth the project risk warrants. That’s the thread running through the whole piece.
CRISPR Off-Target Risk Control: 3-Phase Checklist for Cell Line Editing
Pre-editing checklist: reduce risk before the edit begins
Define the edit goal and choose the lowest-risk editing method
Before you order a single reagent, get very clear on what the edit is meant to do. A knockout, a knock-in, a single-nucleotide change, and transcriptional modulation do not carry the same off-target risk. They also do not call for the same tool.
The broad risk order is straightforward. DSB-forming nucleases such as Cas9 and Cas12 sit at the top end of risk because they can drive large deletions, translocations, and DNA-damage responses [1][7]. Base editors and prime editors use nickases, so they avoid DSBs and cut the risk of structural variation [1][5]. For transcriptional modulation, epigenetic editors such as dCas9 fused to transcriptional modifiers leave the DNA sequence unchanged [1].
The practical rule is simple: use the least genotoxic method that can still deliver the edit you need. For single-nucleotide changes, CBEs or ABEs are a better fit than HDR, which can still introduce indels [3][1]. For substitutions and small insertions or deletions, prime editing often shows lower off-target activity than standard CRISPR-Cas9 [1]. If you have to use a nuclease, pick a high-fidelity variant such as SpCas9-HiFi, eSpCas9, or SpCas9-HF1 [1][6].
Once the editing approach is set, lock down the working cell line and the exact target sequence.
Confirm cell line identity, history, and target locus sequence
If the cell line is misidentified or cross-contaminated, the rest of the workflow starts to wobble. Even a well-designed guide RNA will not rescue bad starting material. Check cell line identity before any editing begins. At the same time, confirm mycoplasma status and record the current passage number, since high-passage cells can shift genomic stability and editing efficiency [1][6].
Just as important, do not lean only on a reference genome. Sequence the exact target locus in the working cell line. That step helps you spot SNPs or indels that may block guide binding or create new off-target sites [1][6].
After that, move into guide design.
Run in silico off-target screening before selecting reagents
Once the target locus is confirmed, screen candidate guide RNAs in silico before you commit to wet-lab work. Use both alignment-based tools, such as Cas-OFFinder or FlashFry, and scoring-based tools, such as CFD scoring or DeepCRISPR. The first group helps find genomic sites with sequence homology. The second helps rank those sites by predicted cleavage probability [1][5].
When guides are being shortlisted, a cleaner off-target profile should beat raw on-target efficiency. A guide with 70% on-target efficiency and no predicted off-targets is a safer place to start than one with 90% efficiency and several high-risk sites [6]. In some settings, shortening guide length from 20 bp to 17-18 bp can cut off-target events by up to 500-fold without much loss of on-target accuracy [5]. Aim for GC content between 40% and 60%, and avoid runs of four or more identical bases [6][5].
That said, in silico screening has limits. It does not account well for chromatin state, cell cycle, or cell-specific context [1][6][4]. Think of it as a filter, not proof. It narrows the field, but it does not replace experimental confirmation.
Bring the highest-risk predicted sites forward into the editing and validation plan.
sbb-itb-ffee270
Editing checklist: control editor choice, delivery, and exposure
Use high-specificity editors and well-ranked guide RNAs
Start with the predicted off-target shortlist and use it to choose the editor. In most cases, a high-fidelity SpCas9 variant - SpCas9-HiFi, eSpCas9, or SpCas9-HF1 - is a better default than wild-type SpCas9 [6][1]. Wild-type SpCas9 can tolerate up to three to five base-pair mismatches, especially in the PAM-distal region, and that creates meaningful off-target risk in sensitive cell lines [3].
A simple rule helps here: use the least active high-fidelity editor that still delivers the intended edit.
For base editors, track bystander edits and RNA off-target effects separately from DNA off-target risk [1][8]. Those are different failure modes, and they need separate checks. If you can make the edit without double-strand breaks, base editing or prime editing may fit better in higher-risk workflows [1][8].
Once the editor is chosen, the next job is to keep its time inside the cell as short as possible.
Limit editor persistence with transient delivery and minimum effective dose
Editor persistence matters just as much as editor choice. The longer the editor stays active in the cell, the more time it has to act at low-probability sites. That makes delivery format a major control point.
Use transient delivery such as RNPs or mRNA, and avoid plasmid DNA or viral vectors that extend editor expression [1][5]. In practice, RNP delivery should be the default [6].
Dose matters too. High nuclease concentration increases the chance of cleavage at low-sensitivity off-target sites [5]. Use the minimum effective dose. If transfection efficiency is poor, don't just throw in more reagent and hope for the best. That often shifts the problem rather than fixing it.
Add precision safeguards for higher-risk workflows
Some workflows need extra guardrails. That is especially true for targets near oncogenes, tumour suppressors, or in p53-sensitive cell lines, where one off-target event can have outsized cost [1][6][3].
Useful safeguards include:
- Paired nickases, which require two nearby cuts. A single off-target nick is usually repaired without mutation, so off-target risk drops a lot compared with a standard nuclease setup [4][1].
- Inducible, light-controlled, or split-Cas9 systems, which help keep editor activity within a tight window when delivery is efficient and exposure must stay brief [1].
- Anti-CRISPR (Acr) proteins, which act as a shutdown switch. These naturally occurring Acr proteins can deactivate the CRISPR-Cas complex after a defined interval, giving you a molecular brake on editor activity [1].
Post-editing checklist: detect off-target events and validate clones
Screen predicted off-target sites with targeted sequencing
Once editing is done, confirm the intended change at the on-target locus first. For a fast first pass in pooled cells, you can use a mismatch-cleavage assay such as T7 Endonuclease I, a restriction digest, or a flanking PCR. Just be careful with interpretation: each of these methods has sensitivity limits, especially for rare edits or homozygous variants [9].
For clone-level validation, targeted amplicon NGS is the standard. It gives you a quantitative view of allele frequency and can detect variants down to 0.01% to 0.1% [3].
Sequence every predicted off-target site with targeted amplicon NGS. That should be the default validation step.
Escalate to genome-wide or structural assays when project risk is higher
Site-by-site screening is not always enough. If the editor, target locus, or cell line suggests hidden risk, move to assays that can pick up events you did not predict in advance.
Genome-wide discovery assays such as GUIDE-seq and CIRCLE-seq do not need prior off-target site lists. GUIDE-seq can detect off-target sites with indel frequencies as low as 0.03% [2]. CIRCLE-seq can identify up to 94% of off-target sites in vitro [3]. These methods are useful when cell-type context may mask off-target activity.
If you are worried about large rearrangements, standard amplicon reads may miss the main problem. Deletions, inversions, and translocations need assays built for structural changes, such as CAST-seq and UDiTaS [1].
Whole genome sequencing (WGS) is the broadest option. It can detect indels, structural variation, and copy-number changes across the genome [1]. The trade-off is depth and cost: it usually needs 20–60× coverage, which makes it a poor fit for routine screening of bulk populations [1].
Use targeted amplicon NGS for predicted sites. Move to genome-wide or structural assays for higher-risk projects. For base or prime editors, add RNA-seq to check RNA-level off-target effects.
Select multiple independent clones and document release criteria
After the sequence checks, test the phenotype in more than one clone.
Do not move ahead with a single edited clone. Isolate and expand at least two to three independent clonal populations and compare them with an unedited parental control [4][9]. Remove clones that show instability or phenotype drift [3]. Then confirm the intended edit at the required allele state, whether heterozygous or homozygous, using targeted amplicon NGS [9].
Documentation is not admin work at the end. It is part of clone release. Record the parental line background, the sgRNA design, the nuclease variant, the delivery method, and all QC results [3]. A clone should only move forward when the intended edit is confirmed, predicted off-target sites are clear, and the full record is in place.
Genome Editing with CRISPR: How to Effectively Minimize Off-Target Effects
Conclusion: a three-phase checklist for cleaner cell line edits
Taken together, the checklist treats off-target control as a staged process, not a one-off QC check. The goal is simple: cut risk early, limit editor activity, then verify the outcome.
Validation depth should match risk. Release only multiple independent clones confirmed at the intended allele state.
FAQs
Why not rely on one clone?
Relying on a single clone is risky. CRISPR editing is not perfectly specific, so it can introduce unintended off-target mutations.
That’s why teams usually expand multiple clonal populations. Doing this makes it easier to find a line that carries the intended on-target edit without harmful off-target changes.
There’s another reason too: cell lines can show genetic heterogeneity. Sequencing multiple clones helps confirm that the intended homozygous knockout or other target-site modification is present across the target loci.
When is amplicon NGS enough?
Amplicon-based next-generation sequencing is often enough when you need a targeted and cost-effective way to confirm potential off-target sites flagged by computational tools or other screening methods.
Whole genome sequencing is still the only way to fully quantify off-target effects. But for many applications, that level of analysis just isn’t needed.
How do I choose the safest editor?
Choose the least active CRISPR nuclease variant that still cuts your on-target site well.
You can’t pick the best variant from prediction alone. The only reliable way is to run a small screen across selected nuclease variants and read out editing with next-generation sequencing.
For cultivated meat R&D, that gives you a practical path forward: start with a short list of variants, then test weaker ones step by step until you find the least active option that still edits the target site efficiently.









