

Batch records are critical for compliance and product safety. They document every step of production, ensuring regulatory standards are met. For cultivated meat producers, maintaining sterility and detailed records is non-negotiable. FDA inspections often highlight issues like missing data, incomplete reviews, and poor corrective actions, which can lead to warnings or disruptions.
Key Takeaways:
- Batch Records: Two types - Master Batch Record (MBR) (the "recipe") and Batch Production Record (BPR) (the "execution").
- Common Issues: Human errors (50% of problems), missing in-process checks, incomplete reviews, and poor CAPA (Corrective and Preventive Action) systems.
- FDA Standards: Adherence to ALCOA+ principles (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available) is mandatory.
- Solutions: Independent audits, electronic batch records, and rigorous supplier verification can minimise errors and improve compliance.
Cultivated meat companies like UPSIDE Foods have set a benchmark by ensuring detailed documentation, input traceability, and rapid corrective measures. By learning from these practices, producers can avoid regulatory pitfalls and maintain high-quality standards.
Comprehensive Guide to Documentation and Record-Keeping for FDA Compliance in Life Sciences
sbb-itb-ffee270
Common Problems in Batch Record Documentation
FDA inspection reports consistently highlight a recurring issue: production record review deviations rank among the top GMP deficiencies cited by regulators [7]. For cultivated meat producers, these shortcomings go beyond mere administrative errors - they jeopardise the ability to demonstrate sustained sterile conditions. These problems appear in several forms, as illustrated in the examples below.
Incomplete Reviews and Non-Conformances
One common issue is the failure of Quality Control Units to thoroughly review batch records. Instead of being an integral part of the release process, reviews often occur reactively - only after a product issue has already emerged [7]. This approach leaves significant gaps in production records.
For example, Davis City Pharmacy received an FDA 483 observation due to batch records missing critical details such as component quantities, operational steps, and personnel initials. Similarly, CAPS was cited for lacking required signatures and reviewer verifications in key entries [3]. These lapses are not isolated incidents; studies show that around 52% of documentation violations escalate when robust bioprocess management systems are absent [3].
"It's not the complexity of the process that triggers citations - it's the inconsistency, incompleteness, and poor oversight." - GXP Auditing & Consulting Services [5]
Missing In-Process Check Records
Another frequent deficiency is the absence of proper documentation for in-process checks. These records are crucial, especially at critical control points in aseptic operations. For instance, Nephron Sterile Compounding Center faced citations for failing to document essential gowning steps and aseptic procedures in their batch records [3]. For cultivated meat producers, where sterility is paramount, such omissions make it impossible to confirm compliance with contamination control measures.
Amphastar was also flagged for failing to investigate or document unexpected yield variances or output discrepancies [3]. The risks of such oversights are stark. In one case, an unidentified pharmaceutical facility in 2024/2025 was found storing completed batch records on open shelving and desks. FDA investigators discovered missing pages, including seven from a single record, and an entire "Synthesis Solution" section absent from another [6].
CAPA and Supplier GMP Verification Failures
Beyond documentation errors, the absence of effective corrective processes and supplier verification further undermines batch record reliability. When production deviations occur without a corresponding Corrective and Preventive Action (CAPA) report, the integrity of batch records is compromised [7]. For example, Eugia Pharma Specialities Limited, inspected between 22 January and 2 February 2024, received an FDA 483 for failing to review discrepancies adequately. Their ineffective CAPA system and incomplete investigations led to repeated production problems, forcing a complete overhaul of their investigation and CAPA procedures [9].
Similarly, during an inspection from 26 September to 25 October 2023, Stokes Healthcare Inc. demonstrated poor discrepancy management. The company failed to extend investigations to all affected batches and delayed completing their analyses [9].
"A discrepancy with no corresponding CAPA or deviation report? That's a compliance failure." - GXP Auditing & Consulting Services [5]
Supplier verification issues add another layer of complexity. Empower Clinic Services LLC was cited during an inspection from 18 July to 5 August 2022 for inadequate quality control procedures, including insufficient supplier qualifications and poor investigation processes [9]. For cultivated meat producers, who rely on growth media, cell lines, and other critical inputs, ensuring supplier GMP compliance is vital to maintaining the integrity of batch records.
FDA Requirements for Batch Records
The FDA's rules for batch records revolve around 21 CFR Part 117, which sets the baseline for food safety. When it comes to cultivated meat, where maintaining sterility during the cell culture phase is crucial, documentation often needs to meet the stricter standards of Part 111 or Part 211, in addition to Part 117 [10][14]. This underscores how precise documentation is essential for ensuring the safety and effectiveness of cultivated meat production.
Core Standards for Batch Records
Each batch requires two key documents:
- Master Batch Record (MBR): The approved template outlining the production process.
- Batch Production Record (BPR): A detailed record of what actually happens during the production run [12][2].
The BPR must include specifics like batch or lot numbers, equipment details, cleaning dates, component identifiers, exact measurements, and comparisons of actual versus theoretical yields [10][14].
"The batch production record must accurately follow the appropriate master manufacturing record and you must perform each step in the production of the batch." – 21 CFR 111.255 [12]
Every critical step must be recorded immediately, with both the performer’s and verifier’s initials noted [10][11]. The FDA requires adherence to ALCOA(+) principles, meaning records must be Attributable, Legible, Contemporaneous, Original, and Accurate - as well as Complete, Consistent, Enduring, and Available [1].
If there’s any deviation from the Master Manufacturing Record, it must be investigated thoroughly. This includes documenting the issue, conducting a root cause analysis, and implementing a Corrective and Preventive Action (CAPA) plan [8][1]. Initial assessments of deviations should be logged within 24–48 hours of detection [8]. For facilities using electronic systems, compliance with 21 CFR Part 11 is mandatory. This includes validated electronic signatures and secure, time-stamped audit trails [8][1].
Record Retention and Review Procedures
Proper record retention and review processes are critical for staying compliant and ensuring product safety. In sterile production, like that of cultivated meat, every detail in the batch records must undergo meticulous review. The Quality Control (QC) team is responsible for reviewing all batch records, monitoring results, and testing data before a batch can be approved for distribution [10][13].
"All drug product production and control records shall be reviewed and approved by the quality control unit before a batch is released or distributed." – 21 CFR 211.192 [2]
Manufacturers often aim to complete 95% of batch reviews within 30 days of production [2]. However, for the more complex sterile processes involved in cultivated meat, reviews typically take 7–10 days, with high-performing facilities achieving times under 7 days [2]. Electronic batch record systems can significantly speed up these reviews , such as those integrated into cultivated meat production systems, - reducing the time by half compared to paper-based methods - as long as they’re validated to meet Part 11 requirements and maintain data integrity [1].
What FDA-Approved Cultivated Meat Companies Did Right
FDA-approved cultivated meat companies have set the bar high by adopting practices that address documentation challenges and meet rigorous safety standards.
When UPSIDE Foods became the first cultivated meat company to pass the FDA's pre-market consultation in November 2022, they established a model for the industry. The FDA issued a "no further questions" letter after thoroughly reviewing their production process, which included cell line establishment, cell banks, manufacturing controls, and all components and inputs [16]. This achievement highlighted the importance of detailed documentation in meeting the FDA's strict requirements.
Meeting Sterility and Compliance Standards
UPSIDE Foods' standout achievement was their exhaustive approach to input traceability. Every production component was carefully documented, ensuring a clear chain of accountability from the initial cell line to the final product [16]. This level of transparency allowed FDA reviewers to trace every step of the manufacturing process, confirming that all safety standards were consistently met.
"The FDA's pre-market consultation with the firm included an evaluation of the firm's production process and the cultured cell material made by the production process, including the establishment of primary and immortalised cell lines and cell banks, manufacturing controls, and all components and inputs." – U.S. Food and Drug Administration [16]
Other successful companies followed suit by implementing detailed aseptic process documentation. This included critical steps like gowning procedures and sterile handling operations [3]. Unlike earlier documentation failures, these companies employed tiered review systems, involving operator checks, production supervision, and quality unit reviews, to catch potential errors before batch release [15]. Electronic batch record systems also played a pivotal role, enforcing mandatory sign-offs at each stage and maintaining immutable audit trails in line with 21 CFR Part 11 requirements [3][2].
These stringent practices naturally extended into how companies handled deviations and failures.
CAPA Processes for Batch Failures
When batches failed to meet specifications, FDA-approved companies took swift and systematic action. Their Corrective and Preventive Action (CAPA) processes included formal root cause analyses, impact assessments, and clearly documented corrective actions [3]. Any deviations were managed within an integrated quality assurance framework, ensuring that all issues were thoroughly investigated, justified, and documented before production continued [2].
Looking ahead, data integrity is set to be a major focus of FDA enforcement actions for 2024–2025 [1].
How to Improve Your Batch Record Practices
Strengthening batch record practices requires precise documentation to address common failures often identified during FDA inspections. Here are some strategies to tackle key challenges.
Conduct Independent Batch Record Audits
Regular third-party audits can uncover issues that internal reviews might overlook. Start by focusing on critical systems like Laboratory Information Management Systems (LIMS), Manufacturing Execution Systems (MES), and Enterprise Resource Planning (ERP). Prioritise documents with high regulatory impact, such as release testing records, stability data, and batch production records.
One effective method is sample retrieval testing. Randomly select recent batches and reconstruct their production and laboratory history. This can help pinpoint missing data, incomplete signatures, or documentation gaps that could lead to regulatory citations. Cross-check system-generated audit trails with manual entries to identify unauthorised changes or deletions.
Review all Out-of-Specification (OOS) and Out-of-Trend (OOT) reports from the past year. Evaluate whether root cause analyses were thorough and if Corrective and Preventive Actions (CAPAs) were adequately implemented. It's worth noting that documentation issues account for 21% of FDA warning letters, while human error contributes to 50% of batch record problems in pharmaceutical manufacturing [2].
"It's not the complexity of the process that triggers citations - it's the inconsistency, incompleteness, and poor oversight." – GXP Auditing & Consulting Services [5]
Simulate regulatory inspections through periodic mock reviews. This practice helps teams recognise inconsistencies and potential data integrity issues before an actual audit. Ensure all records follow ALCOA+ principles: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available.
Once documentation integrity is solid, focus on verifying the quality of all production inputs.
Test All Inputs for Microbiological Contamination
Independent sterility and potency testing for all inputs is essential - don’t rely solely on supplier Certificates of Analysis (CoAs). This is particularly crucial for cultivated meat producers, as contamination can jeopardise entire batches.
For instance, in February 2013, Central Admixture Pharmacy Services faced FDA citations due to inadequate microbial control during batch release of sterile products. The company had to introduce detailed microbial control procedures in its Standard Operating Procedures (SOPs) [4].
In-process microbial checkpoints can prevent over-reliance on final product testing. Incorporate these checkpoints into batch release SOPs and maintain strict contemporaneous documentation. Record all test results and manufacturing steps as they happen to avoid back-dating or delayed entries, which could result in FDA citations.
Keep comprehensive supplier files, including CoAs, audit reports, quality agreements, and a history of any deviations related to incoming materials.
Strengthening batch record practices further involves aligning processes with established standards like HACCP and GCCP.
Align Records with HACCP and GCCP Standards
Incorporating Hazard Analysis Critical Control Point (HACCP) principles into batch records ensures critical process variables are monitored and documented throughout production. This includes establishing in-process microbial testing checkpoints rather than relying solely on final-stage tests.
For cultivated meat producers, adherence to Good Cell Culture Practice (GCCP) standards is equally vital. Batch records should include details of aseptic manipulations, gowning procedures, and environmental monitoring tied to batch release criteria [3][4]. These steps help maintain compliance and ensure product safety.
Industry data shows that 52% of documentation violations escalate when proper batch manufacturing software is not in place [3][4]. A case in point: in February 2023, Nephron Sterile Compounding Centre received an FDA observation due to the absence of control procedures for verifying critical process variables before batch release [4]. This highlights the need for proactive documentation aligned with recognised standards.
Transitioning to Electronic Batch Records (EBR) can significantly reduce documentation errors - by as much as 50% - through real-time data collection and automated workflows [2]. These systems flag missing microbial test results or incomplete reviews before a batch advances, minimising human error.
"The FDA expects records to be ALCOA(+): Attributable, Legible, Contemporaneous, Original, Accurate - plus Complete, Consistent, Enduring, and Available." – Atlas Compliance [1]
Every unexplained discrepancy or deviation in batch records should be linked to a formal investigation and CAPA system. Limit write and delete permissions to protect the integrity of electronic microbiological test data. Competitive manufacturers aim to review and release 95% of batches within 30 days of production completion [2].
These actions not only reduce the risk of citations but also align with the rigorous documentation standards highlighted in recent FDA inspections.
Biopharma vs Cultivated Meat: Batch Record Differences
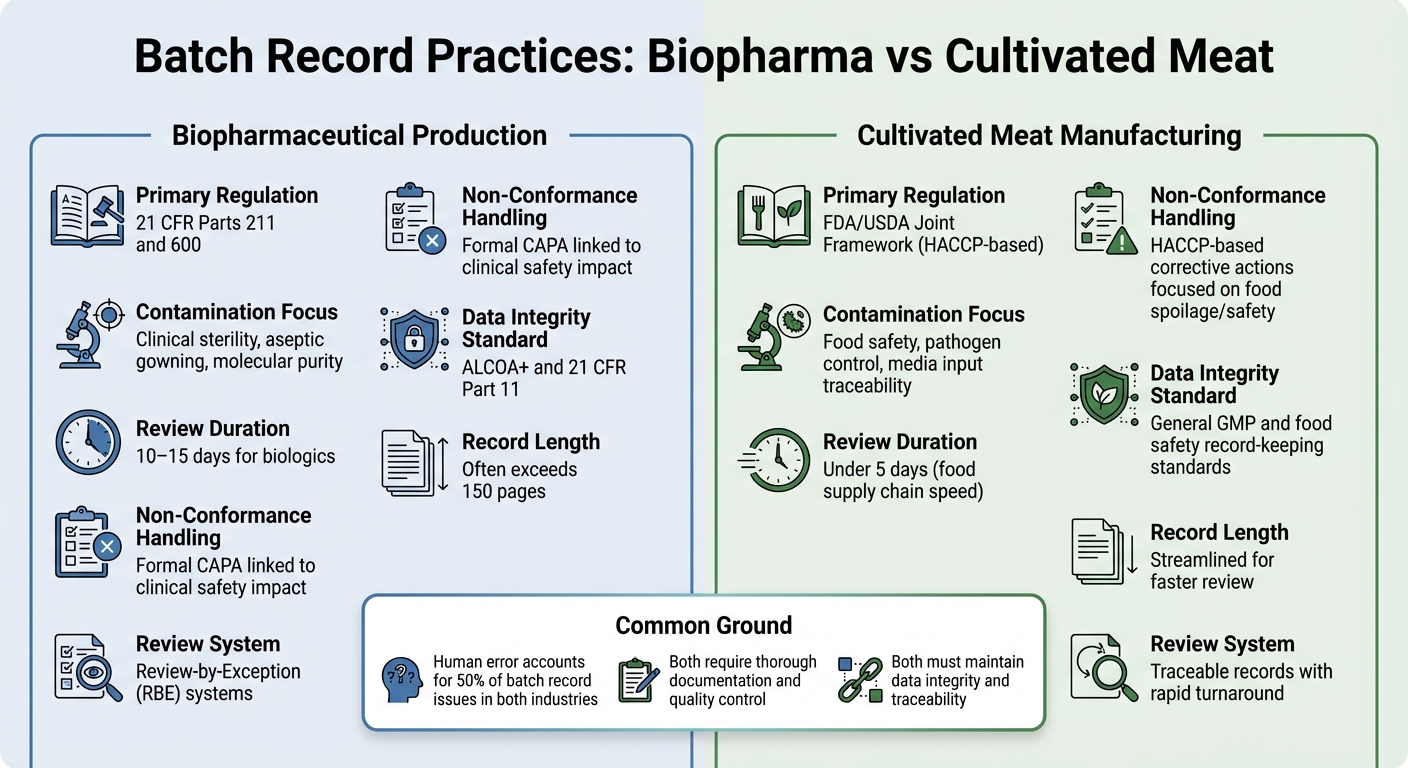
Biopharmaceutical vs Cultivated Meat Batch Record Requirements Comparison
Looking at the differences in batch record practices between biopharmaceutical production and cultivated meat manufacturing offers a clearer picture of how regulatory demands shape documentation priorities in these industries.
Both sectors require thorough documentation, but their regulatory frameworks and control objectives differ significantly. In biopharma, batch records are tightly regulated under 21 CFR Parts 211 and 600, which require the Quality Control unit to review and approve all production and control records before a batch can be released [2]. Cultivated meat producers, on the other hand, typically follow HACCP and GCCP standards. These focus more on food safety and pathogen control, rather than the clinical-grade sterility demanded for injectable biologics.
Biopharma batch records are often extensive, sometimes exceeding 150 pages, and the review process can take 10–15 days. To streamline this, many biopharma companies use Review-by-Exception (RBE) systems, which summarise key deviations into a compact report. Meanwhile, cultivated meat producers aim for traceable records that can be reviewed in less than five days, reflecting the faster pace of the food supply chain [2].
The content of these records also highlights the differing priorities. Biopharma inspections often focus on aseptic processing details, such as gowning procedures and environmental controls. In contrast, cultivated meat records must emphasise media inputs and microbiological testing to ensure food safety. For cultivated meat, the challenge lies in tracking complex media inputs, documenting microbiological tests for all materials, and meeting food safety critical limits - without adhering to the stricter sterility requirements of pharmaceuticals.
Contamination and Non-Conformance Trends
| Feature | Biopharmaceutical Production | Cultivated Meat Manufacturing |
|---|---|---|
| Primary Regulation | 21 CFR Parts 211 and 600 [2] | FDA/USDA Joint Framework (HACCP-based) |
| Contamination Focus | Clinical sterility, aseptic gowning, molecular purity [2] | Food safety, pathogen control, media input traceability |
| Review Duration | 10–15 days for biologics [2] | Under 5 days (food supply chain speed) |
| Non-Conformance Handling | Formal CAPA linked to clinical safety impact [2] | HACCP-based corrective actions focused on food spoilage/safety |
| Data Integrity Standard | ALCOA+ and 21 CFR Part 11 [1] | General GMP and food safety record-keeping standards |
While human error rates are similar in both industries - around 50% of batch record issues arise from human mistakes [2] - the stakes are different. In biopharma, even a single undocumented deviation could have serious implications for patient safety. For cultivated meat, contamination risks are more about foodborne pathogens and spoilage, which can affect entire production runs.
Conclusion
Batch records serve as the official log for every cultivated meat production run - if a step isn't recorded, regulators consider it not performed [6][3]. This highlights the importance of precise documentation and strict quality control.
FDA inspections emphasise that data integrity must align with ALCOA+ principles [1]. Quality Control teams are required to review and approve all production records before a batch can be released [2][17], and any deviations must be promptly investigated with a documented root cause analysis [2][5]. While human error accounts for 50% of batch record issues, dual-level reviews and structured CAPA (Corrective and Preventive Action) processes can help reduce these risks [2][5].
"It's not the complexity of the process that triggers citations - it's the inconsistency, incompleteness, and poor oversight." - GXP Auditing & Consulting Services [5]
To overcome these challenges, cultivated meat producers should focus on independent audits, rigorous testing of food-safe ingredients for microbiological contamination, and ensuring documentation adheres to HACCP and GCCP standards. Implementing electronic batch record systems, validated under 21 CFR Part 11 [1], can significantly minimise errors and speed up review processes.
The regulatory environment demands precision, but it is navigable. By learning from the errors of biopharma - such as missing signatures at Qinhuangdao Zizhu Pharmaceutical [17], insufficient dual verification at Terumo Corp [18], and inadequate deviation documentation at Torrent Pharmaceuticals [18] - cultivated meat companies can establish compliant systems from the outset. Incorporating these lessons enables proactive compliance and consistent quality. Secure record retention, timely deviation reporting, and conducting realistic mock audits will ensure batch records remain inspection-ready and production runs are fully traceable.
For more resources and expert guidance on maintaining high production standards in cultivated meat manufacturing, visit Cellbase.
FAQs
What should a batch record include for cultivated meat?
A batch record for cultivated meat serves as a comprehensive log of the entire manufacturing process. It must include detailed processing instructions, step-by-step execution records, and note any deviations that occur during production. Additionally, it should document in-process testing and release testing to confirm the product meets safety, quality, and regulatory standards.
How can we prove sterility using batch records?
Proving sterility through batch records involves thoroughly examining documented sterilisation procedures, testing outcomes, and media quality control reports to ensure they meet regulatory requirements. It's crucial to address any deviations or failed tests through detailed investigations and CAPAs (Corrective and Preventive Actions). This process ensures that every step has been adhered to and any issues have been properly resolved to uphold sterility standards.
When are electronic batch records (Part 11) required?
Electronic batch records are essential under Part 11 when electronic systems are used to document, investigate, and justify batch record deviations. They play a critical role in ensuring compliance with 21 CFR Part 211.192, safeguarding data integrity, meeting investigation timelines, and ensuring effective management oversight.









