

Chargenprotokolle sind entscheidend für die Einhaltung von Vorschriften und die Produktsicherheit. Sie dokumentieren jeden Produktionsschritt und stellen sicher, dass regulatorische Standards eingehalten werden. Für Produzenten von kultiviertem Fleisch ist die Aufrechterhaltung der Sterilität und detaillierte Aufzeichnungen unverzichtbar. FDA-Inspektionen heben oft Probleme wie fehlende Daten, unvollständige Überprüfungen und mangelhafte Korrekturmaßnahmen hervor, die zu Warnungen oder Unterbrechungen führen können.
Wichtige Erkenntnisse:
- Chargenprotokolle: Zwei Typen - Master Batch Record (MBR) (das "Rezept") und Batch Production Record (BPR) (die "Ausführung").
- Häufige Probleme: Menschliche Fehler (50% der Probleme), fehlende In-Prozess-Kontrollen, unvollständige Überprüfungen und mangelhafte CAPA-Systeme (Corrective and Preventive Action).
- FDA-Standards: Die Einhaltung der ALCOA+-Prinzipien (Attributable, Lesbar, Zeitnah, Original, Genau, Vollständig, Konsistent, Dauerhaft, Verfügbar) ist obligatorisch.
- Lösungen: Unabhängige Audits, elektronische Chargenprotokolle und strenge Lieferantenüberprüfungen können Fehler minimieren und die Einhaltung verbessern.
Kultivierte Fleischunternehmen wie UPSIDE Foods haben einen Maßstab gesetzt, indem sie eine detaillierte Dokumentation, Rückverfolgbarkeit der Eingaben und schnelle Korrekturmaßnahmen sicherstellen. Durch das Lernen aus diesen Praktiken können Produzenten regulatorische Fallstricke vermeiden und hohe Qualitätsstandards aufrechterhalten.
Umfassender Leitfaden zur Dokumentation und Aufzeichnung für FDA-Konformität in den Lebenswissenschaften
sbb-itb-ffee270
Häufige Probleme bei der Chargenprotokolldokumentation
FDA-Inspektionsberichte heben konsequent ein wiederkehrendes Problem hervor: Abweichungen bei der Überprüfung von Produktionsaufzeichnungen gehören zu den am häufigsten von Regulierungsbehörden zitierten GMP-Mängeln [7]. Für Produzenten von kultiviertem Fleisch gehen diese Mängel über bloße Verwaltungsfehler hinaus - sie gefährden die Fähigkeit, anhaltend sterile Bedingungen nachzuweisen. Diese Probleme treten in verschiedenen Formen auf, wie in den folgenden Beispielen veranschaulicht.
Unvollständige Überprüfungen und Nichtkonformitäten
Ein häufiges Problem ist das Versäumnis der Qualitätssicherungseinheiten, Chargenprotokolle gründlich zu überprüfen. Anstatt ein integraler Bestandteil des Freigabeprozesses zu sein, erfolgen Überprüfungen oft reaktiv - erst nachdem ein Produktproblem bereits aufgetreten ist [7]. Dieser Ansatz lässt erhebliche Lücken in den Produktionsaufzeichnungen.
Zum Beispiel erhielt Davis City Pharmacy eine FDA 483-Beobachtung aufgrund fehlender kritischer Details in den Chargenprotokollen, wie Komponentenmengen, Arbeitsschritte und Initialen des Personals. Ebenso wurde CAPS wegen fehlender erforderlicher Unterschriften und Überprüfungsbestätigungen in wichtigen Einträgen zitiert [3]. Diese Versäumnisse sind keine Einzelfälle; Studien zeigen, dass etwa 52% der Dokumentationsverstöße eskalieren, wenn robuste Bioprozess-Managementsysteme fehlen [3].
"Es ist nicht die Komplexität des Prozesses, die zu Beanstandungen führt - es ist die Inkonsistenz, Unvollständigkeit und schlechte Aufsicht." - GXP Auditing & Consulting Services [5]
Fehlende In-Prozess-Kontrollaufzeichnungen
Ein weiterer häufiger Mangel ist das Fehlen ordnungsgemäßer Dokumentation für In-Prozess-Kontrollen. Diese Aufzeichnungen sind besonders an kritischen Kontrollpunkten in aseptischen Operationen entscheidend. Zum Beispiel erhielt Nephron Sterile Compounding Center Beanstandungen, weil wesentliche Bekleidungsschritte und aseptische Verfahren in ihren Chargenaufzeichnungen nicht dokumentiert wurden [3]. Für Produzenten von kultiviertem Fleisch, bei denen Sterilität von größter Bedeutung ist, machen solche Auslassungen es unmöglich, die Einhaltung von Kontaminationskontrollmaßnahmen.
zu bestätigen.Amphastar wurde auch dafür gerügt, dass es versäumt hat, unerwartete Ertragsabweichungen oder Produktionsdiskrepanzen zu untersuchen oder zu dokumentieren [3]. Die Risiken solcher Versäumnisse sind gravierend. In einem Fall wurde festgestellt, dass eine nicht identifizierte pharmazeutische Einrichtung in den Jahren 2024/2025 abgeschlossene Chargenprotokolle in offenen Regalen und auf Schreibtischen lagerte. FDA-Ermittler entdeckten fehlende Seiten, darunter sieben aus einem einzigen Protokoll, und einen gesamten Abschnitt "Syntheselösung", der in einem anderen fehlte [6].
CAPA und Lieferanten-GMP-Verifizierungsfehler
Über Dokumentationsfehler hinaus untergräbt das Fehlen effektiver Korrekturprozesse und Lieferantenverifizierung weiter die Zuverlässigkeit von Chargenprotokollen.Wenn Produktionsabweichungen ohne einen entsprechenden Bericht zur Korrektur- und Vorbeugemaßnahme (CAPA) auftreten, wird die Integrität der Chargendokumentation beeinträchtigt [7]. Zum Beispiel, Eugia Pharma Specialities Limited, zwischen dem 22. Januar und dem 2. Februar 2024 inspiziert, erhielt eine FDA 483 wegen unzureichender Überprüfung von Abweichungen. Ihr ineffektives CAPA-System und unvollständige Untersuchungen führten zu wiederholten Produktionsproblemen, was eine vollständige Überarbeitung ihrer Untersuchungs- und CAPA-Verfahren erforderte [9].
Ähnlich zeigte Stokes Healthcare Inc. während einer Inspektion vom 26. September bis 25. Oktober 2023 ein schlechtes Abweichungsmanagement. Das Unternehmen versäumte es, Untersuchungen auf alle betroffenen Chargen auszuweiten und verzögerte den Abschluss ihrer Analysen [9].
"Eine Abweichung ohne entsprechenden CAPA- oder Abweichungsbericht? Das ist ein Compliance-Fehler." - GXP-Audits & Beratungsdienste [5]
Probleme bei der Lieferantenverifizierung fügen eine weitere Ebene der Komplexität hinzu. Empower Clinic Services LLC wurde während einer Inspektion vom 18. Juli bis 5. August 2022 wegen unzureichender Qualitätskontrollverfahren, einschließlich unzureichender Lieferantenqualifikationen und mangelhafter Untersuchungsprozesse, zitiert [9]. Für Produzenten von kultiviertem Fleisch, die auf Wachstumsmedien, Zelllinien und andere kritische Inputs angewiesen sind, ist die Sicherstellung der GMP-Konformität der Lieferanten entscheidend für die Aufrechterhaltung der Integrität der Chargenprotokolle.
FDA-Anforderungen für Chargenprotokolle
Die FDA-Vorschriften für Chargenprotokolle drehen sich um 21 CFR Teil 117, welches den Grundstein für Lebensmittelsicherheit legt.Wenn es um kultiviertes Fleisch geht, bei dem die Aufrechterhaltung der Sterilität während der Zellkulturphase entscheidend ist, muss die Dokumentation oft den strengeren Standards von Teil 111 oder Teil 211, zusätzlich zu Teil 117 [10][14]. entsprechen. Dies unterstreicht, wie präzise Dokumentation entscheidend für die Sicherheit und Wirksamkeit der Produktion von kultiviertem Fleisch ist.
Kernstandards für Chargenprotokolle
Jede Charge erfordert zwei wichtige Dokumente:
- Master Batch Record (MBR): Die genehmigte Vorlage, die den Produktionsprozess umreißt.
- Batch Production Record (BPR): Ein detailliertes Protokoll darüber, was während des Produktionslaufs tatsächlich passiert [12][2].
Der BPR muss spezifische Angaben wie Chargen- oder Losnummern, Gerätedetails, Reinigungsdaten, Komponentenkennungen, genaue Messungen und Vergleiche von tatsächlichen gegenüber theoretischen Ausbeuten enthalten [10][14].
"Der Chargenproduktionsbericht muss dem entsprechenden Master-Herstellungsbericht genau folgen, und Sie müssen jeden Schritt bei der Produktion der Charge durchführen." – 21 CFR 111.255 [12]
Jeder kritische Schritt muss sofort aufgezeichnet werden, wobei sowohl die Initialen des Ausführenden als auch des Prüfers notiert werden [10][11]. Die FDA verlangt die Einhaltung der ALCOA(+) Prinzipien, was bedeutet, dass Aufzeichnungen Zurechenbar, Lesbar, Zeitnah, Original und Genau - sowie Vollständig, Konsistent, Dauerhaft und Verfügbar sein müssen [1].
Wenn es Abweichungen vom Master Manufacturing Record gibt, müssen diese gründlich untersucht werden. Dazu gehört die Dokumentation des Problems, die Durchführung einer Ursachenanalyse und die Implementierung eines Corrective and Preventive Action (CAPA)-Plans [8] [1]. Erste Bewertungen von Abweichungen sollten innerhalb von 24–48 Stunden nach Entdeckung protokolliert werden [8]. Für Einrichtungen, die elektronische Systeme verwenden, ist die Einhaltung von 21 CFR Part 11 obligatorisch. Dies umfasst validierte elektronische Signaturen und sichere, zeitgestempelte Prüfpfade [8] [1].
Verfahren zur Aufbewahrung und Überprüfung von Aufzeichnungen
Ordnungsgemäße Verfahren zur Aufbewahrung und Überprüfung von Aufzeichnungen sind entscheidend, um konform zu bleiben und die Produktsicherheit zu gewährleisten.In der sterilen Produktion, wie der von kultiviertem Fleisch, muss jedes Detail in den Chargenprotokollen einer sorgfältigen Überprüfung unterzogen werden. Das Qualitätskontrollteam (QC) ist verantwortlich für die Überprüfung aller Chargenprotokolle, die Überwachung der Ergebnisse und die Testdaten, bevor eine Charge zur Verteilung freigegeben werden kann [10] [13].
"Alle Produktions- und Kontrollaufzeichnungen von Arzneimitteln müssen von der Qualitätskontrolleinheit überprüft und genehmigt werden, bevor eine Charge freigegeben oder verteilt wird." – 21 CFR 211.192 [2]
Hersteller streben oft an, 95% der Chargenüberprüfungen innerhalb von 30 Tagen nach der Produktion abzuschließen [2] . Jedoch dauern die Überprüfungen für die komplexeren sterilen Prozesse, die bei kultiviertem Fleisch beteiligt sind, typischerweise 7–10 Tage, wobei leistungsstarke Einrichtungen Zeiten unter 7 Tagen erreichen [2]. Elektronische Chargenaufzeichnungssysteme können diese Überprüfungen erheblich beschleunigen, wie z.B. die in kultivierte Fleischproduktionssysteme, integrierten - wodurch die Zeit im Vergleich zu papierbasierten Methoden halbiert wird - solange sie validiert sind, um die Anforderungen von Teil 11 zu erfüllen und die Datenintegrität zu wahren [1].
Was von der FDA zugelassene Unternehmen für kultiviertes Fleisch richtig gemacht haben
Von der FDA zugelassene Unternehmen für kultiviertes Fleisch haben die Messlatte hoch gelegt, indem sie Praktiken übernommen haben, die Dokumentationsherausforderungen adressieren und strenge Sicherheitsstandards erfüllen.
Als UPSIDE Foods im November 2022 als erstes Unternehmen für kultiviertes Fleisch die Vorab-Marktberatung der FDA bestand, schufen sie ein Modell für die Branche.Die FDA stellte nach einer gründlichen Überprüfung ihres Produktionsprozesses, der die Etablierung von Zelllinien, Zellbanken, Herstellungssteuerungen und alle Komponenten und Eingaben umfasste, ein "keine weiteren Fragen"-Schreiben aus [16]. Diese Errungenschaft unterstrich die Bedeutung detaillierter Dokumentation zur Erfüllung der strengen Anforderungen der FDA.
Erfüllung von Sterilitäts- und Compliance-Standards
Die herausragende Leistung von UPSIDE Foods war ihr umfassender Ansatz zur Rückverfolgbarkeit von Eingaben. Jede Produktionskomponente wurde sorgfältig dokumentiert, um eine klare Verantwortlichkeitskette von der anfänglichen Zelllinie bis zum Endprodukt sicherzustellen [16]. Dieses Maß an Transparenz ermöglichte es den FDA-Prüfern, jeden Schritt des Herstellungsprozesses nachzuvollziehen und zu bestätigen, dass alle Sicherheitsstandards konsequent eingehalten wurden.
"Die Vorabkonsultation der FDA mit dem Unternehmen umfasste eine Bewertung des Produktionsprozesses des Unternehmens und des durch den Produktionsprozess hergestellten kultivierten Zellmaterials, einschließlich der Einrichtung von primären und immortalisierten Zelllinien und Zellbanken, Herstellungssteuerungen sowie aller Komponenten und Eingaben." – U.S. Food and Drug Administration [16]
Andere erfolgreiche Unternehmen folgten diesem Beispiel, indem sie detaillierte aseptische Prozessdokumentationen implementierten. Dazu gehörten kritische Schritte wie Anziehverfahren und sterile Handhabungsoperationen [3]. Im Gegensatz zu früheren Dokumentationsfehlern setzten diese Unternehmen gestufte Überprüfungssysteme ein, die Bedienerprüfungen, Produktionsüberwachung und Qualitätskontrollen umfassten, um potenzielle Fehler vor der Chargenfreigabe zu erkennen [15]. Elektronische Chargenaufzeichnungssysteme spielten ebenfalls eine entscheidende Rolle, indem sie obligatorische Freigaben in jeder Phase durchsetzten und unveränderliche Prüfpfade gemäß den Anforderungen von 21 CFR Part 11 aufrechterhielten [3][2].
Diese strengen Praktiken erstreckten sich natürlich darauf, wie Unternehmen mit Abweichungen und Fehlern umgingen.
CAPA-Prozesse für Chargenfehler
Wenn Chargen die Spezifikationen nicht erfüllten, ergriffen von der FDA zugelassene Unternehmen schnelle und systematische Maßnahmen. Ihre Korrektur- und Vorbeugemaßnahmen (CAPA) umfassten formale Ursachenanalysen, Auswirkungenbewertungen und klar dokumentierte Korrekturmaßnahmen [3]. Alle Abweichungen wurden innerhalb eines integrierten Qualitätssicherungsrahmens verwaltet, um sicherzustellen, dass alle Probleme gründlich untersucht, gerechtfertigt und dokumentiert wurden, bevor die Produktion fortgesetzt wurde [2].
In die Zukunft blickend, wird die Datenintegrität ein Hauptfokus der FDA-Durchsetzungsmaßnahmen für 2024–2025 sein [1].
Wie Sie Ihre Chargenprotokollpraktiken verbessern können
Die Stärkung der Chargenprotokollpraktiken erfordert eine präzise Dokumentation, um häufige Fehler zu beheben, die bei FDA-Inspektionen oft identifiziert werden. Hier sind einige Strategien zur Bewältigung der wichtigsten Herausforderungen.
Unabhängige Chargenprotokoll-Audits durchführen
Regelmäßige Audits durch Dritte können Probleme aufdecken, die interne Überprüfungen möglicherweise übersehen. Beginnen Sie mit der Fokussierung auf kritische Systeme wie Laborinformationsmanagementsysteme (LIMS), Fertigungsausführungssysteme (MES) und Enterprise Resource Planning (ERP). Priorisieren Sie Dokumente mit hoher regulatorischer Auswirkung, wie Freigabetestprotokolle, Stabilitätsdaten und Chargenproduktionsprotokolle.
Eine effektive Methode ist das Testen der Probenentnahme.Zufällig ausgewählte Chargen der letzten Zeit und deren Produktions- und Laborhistorie rekonstruieren. Dies kann helfen, fehlende Daten, unvollständige Unterschriften oder Dokumentationslücken zu identifizieren, die zu regulatorischen Beanstandungen führen könnten. Systemgenerierte Prüfpfade mit manuellen Einträgen abgleichen, um unautorisierte Änderungen oder Löschungen zu erkennen.
Alle Berichte über Abweichungen (Out-of-Specification, OOS) und Trends (Out-of-Trend, OOT) des letzten Jahres überprüfen. Bewerten, ob die Ursachenanalysen gründlich waren und ob Korrektur- und Vorbeugemaßnahmen (CAPAs) angemessen umgesetzt wurden. Es ist erwähnenswert, dass Dokumentationsprobleme 21 % der FDA-Warnschreiben ausmachen, während menschliches Versagen zu 50 % der Chargenproblems in der pharmazeutischen Herstellung beiträgt [2].
"Es ist nicht die Komplexität des Prozesses, die zu Beanstandungen führt - es ist die Inkonsistenz, Unvollständigkeit und schlechte Aufsicht." – GXP-Auditierung & Beratungsdienste [5]
Simulieren Sie behördliche Inspektionen durch regelmäßige Scheinüberprüfungen. Diese Praxis hilft Teams, Inkonsistenzen und potenzielle Datenintegritätsprobleme vor einem tatsächlichen Audit zu erkennen. Stellen Sie sicher, dass alle Aufzeichnungen den ALCOA+-Prinzipien folgen: Attributierbar, Lesbar, Zeitnah, Original, Genau, Vollständig, Konsistent, Dauerhaft und Verfügbar.
Sobald die Dokumentationsintegrität gesichert ist, konzentrieren Sie sich auf die Überprüfung der Qualität aller Produktionsinputs.
Testen Sie alle Inputs auf mikrobiologische Kontamination
Unabhängige Sterilitäts- und Wirksamkeitstests für alle Inputs sind unerlässlich - verlassen Sie sich nicht ausschließlich auf Lieferantenanalysenzertifikate (CoAs). Dies ist besonders wichtig für Produzenten von kultiviertem Fleisch, da Kontaminationen ganze Chargen gefährden können.
Zum Beispiel erhielt Central Admixture Pharmacy Services im Februar 2013 FDA-Zitationen aufgrund unzureichender mikrobieller Kontrolle während der Chargenfreigabe steriler Produkte. Das Unternehmen musste detaillierte mikrobiologische Kontrollverfahren in seine Standardarbeitsanweisungen (SOPs) einführen [4].
In-Prozess-mikrobielle Kontrollpunkte können eine übermäßige Abhängigkeit von der Endproduktprüfung verhindern. Integrieren Sie diese Kontrollpunkte in die SOPs zur Chargenfreigabe und führen Sie eine strikte gleichzeitige Dokumentation. Zeichnen Sie alle Testergebnisse und Herstellungsschritte auf, während sie stattfinden, um Rückdatierungen oder verzögerte Einträge zu vermeiden, die zu FDA-Zitationen führen könnten.
Führen Sie umfassende Lieferantenakten, einschließlich CoAs, Auditberichte, Qualitätsvereinbarungen und einer Historie aller Abweichungen im Zusammenhang mit eingehenden Materialien.
Die Stärkung der Chargenaufzeichnungspraktiken beinhaltet auch die Ausrichtung der Prozesse an etablierten Standards wie HACCP und GCCP.
Abgleich von Aufzeichnungen mit HACCP- und GCCP-Standards
Die Einbeziehung der Prinzipien des Hazard Analysis Critical Control Point (HACCP) in Chargendokumentationen stellt sicher, dass kritische Prozessvariablen während der Produktion überwacht und dokumentiert werden. Dies beinhaltet die Einrichtung von mikrobiologischen Testkontrollpunkten im Prozess, anstatt sich ausschließlich auf Endstufentests zu verlassen.
Für Produzenten von kultiviertem Fleisch ist die Einhaltung der Good Cell Culture Practice (GCCP)-Standards ebenso wichtig. Chargendokumentationen sollten Details zu aseptischen Manipulationen, Bekleidungsverfahren und Umweltüberwachung enthalten, die an die Kriterien zur Chargenfreigabe gebunden sind [3][4]. Diese Schritte helfen, die Einhaltung der Vorschriften zu gewährleisten und die Produktsicherheit sicherzustellen.
Branchendaten zeigen, dass 52% der Dokumentationsverstöße zunehmen, wenn keine geeignete Chargenfertigungssoftware vorhanden ist [3][4]. Ein Beispiel: Im Februar 2023 erhielt das Nephron Sterile Compounding Centre eine FDA-Beobachtung aufgrund des Fehlens von Kontrollverfahren zur Überprüfung kritischer Prozessvariablen vor der Chargenfreigabe [4]. Dies unterstreicht die Notwendigkeit einer proaktiven Dokumentation, die mit anerkannten Standards übereinstimmt.
Der Übergang zu elektronischen Chargenprotokollen (EBR) kann Dokumentationsfehler erheblich reduzieren - um bis zu 50% - durch Echtzeit-Datenerfassung und automatisierte Workflows [2]. Diese Systeme kennzeichnen fehlende mikrobiologische Testergebnisse oder unvollständige Überprüfungen, bevor eine Charge weitergeht, und minimieren so menschliche Fehler.
"Die FDA erwartet, dass Aufzeichnungen ALCOA(+) sind: Attributierbar, Lesbar, Zeitnah, Original, Genau - plus Vollständig, Konsistent, Dauerhaft und Verfügbar." – Atlas Compliance [1]
Jede unerklärliche Abweichung oder Diskrepanz in Chargenprotokollen sollte mit einem formellen Untersuchungs- und CAPA-System verknüpft werden. Beschränken Sie Schreib- und Löschberechtigungen, um die Integrität elektronischer mikrobiologischer Testdaten zu schützen. Wettbewerbsfähige Hersteller streben an, 95 % der Chargen innerhalb von 30 Tagen nach Produktionsabschluss zu überprüfen und freizugeben [2].
Diese Maßnahmen reduzieren nicht nur das Risiko von Beanstandungen, sondern entsprechen auch den strengen Dokumentationsstandards, die in den jüngsten FDA-Inspektionen hervorgehoben wurden.
Biopharma vs Cultivated Meat: Unterschiede in den Chargenprotokollen
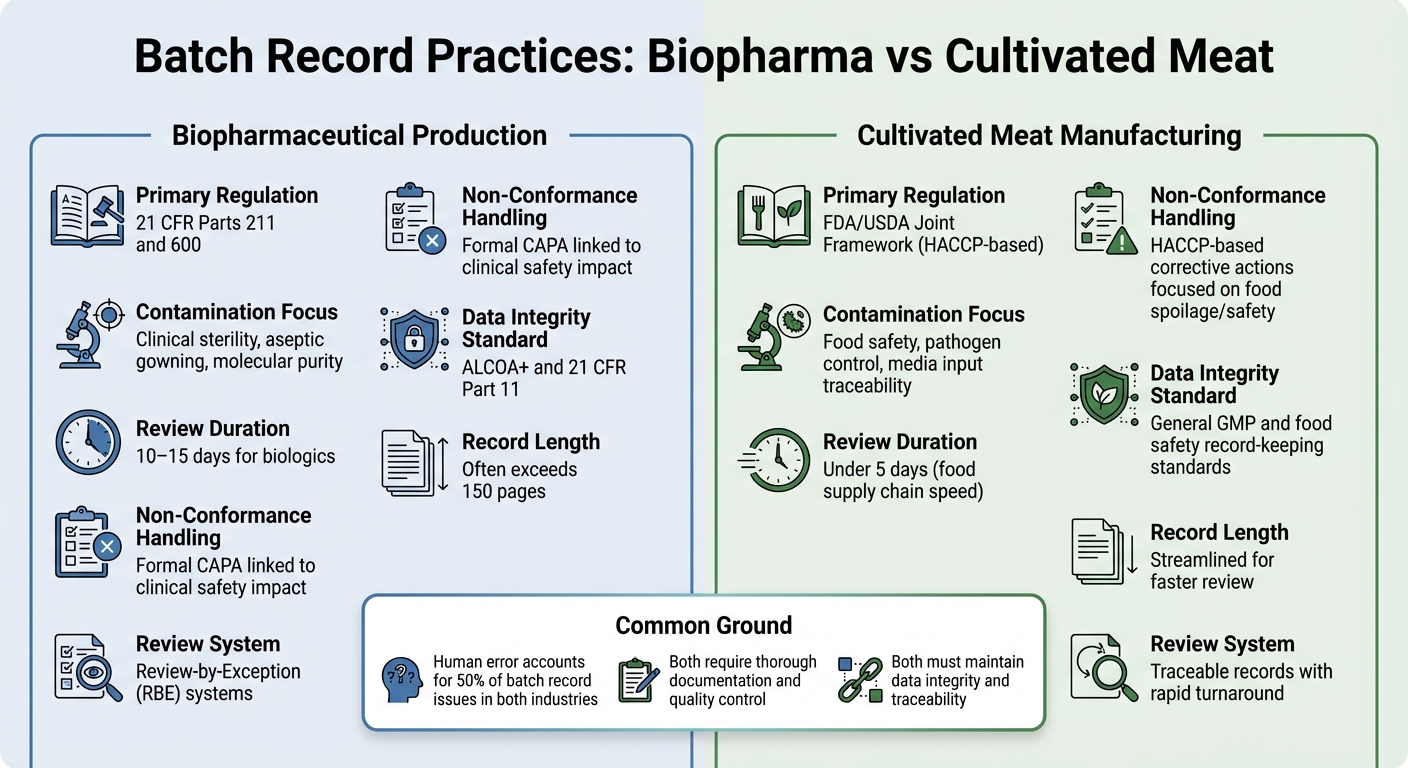
Vergleich der Anforderungen an Chargenprotokolle in der Biopharmazeutik vs. kultiviertem Fleisch
Ein Blick auf die Unterschiede in den Praktiken der Chargenprotokollierung zwischen der biopharmazeutischen Produktion und der Herstellung von kultiviertem Fleisch bietet ein klareres Bild davon, wie regulatorische Anforderungen die Dokumentationsprioritäten in diesen Branchen prägen.
Beide Sektoren erfordern eine gründliche Dokumentation, aber ihre regulatorischen Rahmenbedingungen und Kontrollziele unterscheiden sich erheblich. In der Biopharmazeutik sind Chargenprotokolle streng reguliert unter 21 CFR Teile 211 und 600, die verlangen, dass die Qualitätskontrolleinheit alle Produktions- und Kontrollaufzeichnungen überprüft und genehmigt, bevor eine Charge freigegeben werden kann [2]. Kultivierte Fleischproduzenten hingegen folgen typischerweise den Standards HACCP und GCCP. Diese konzentrieren sich mehr auf Lebensmittelsicherheit und Erregerkontrolle, anstatt auf die klinische Sterilität, die für injizierbare Biologika gefordert wird.
Biopharma-Chargenprotokolle sind oft umfangreich und können manchmal über 150 Seiten umfassen, wobei der Überprüfungsprozess 10–15 Tage dauern kann. Um dies zu optimieren, verwenden viele Biopharma-Unternehmen Review-by-Exception (RBE)-Systeme, die wesentliche Abweichungen in einem kompakten Bericht zusammenfassen. Währenddessen streben Produzenten von kultiviertem Fleisch nach nachvollziehbaren Aufzeichnungen, die in weniger als fünf Tagen überprüft werden können, was das schnellere Tempo der Lebensmittelversorgungskette widerspiegelt [2].
Der Inhalt dieser Aufzeichnungen hebt auch die unterschiedlichen Prioritäten hervor. Biopharma-Inspektionen konzentrieren sich oft auf Details der aseptischen Verarbeitung, wie Bekleidungsverfahren und Umweltkontrollen. Im Gegensatz dazu müssen Aufzeichnungen über kultiviertes Fleisch die Medieninputs und mikrobiologischen Tests betonen, um die Lebensmittelsicherheit zu gewährleisten.Für kultiviertes Fleisch besteht die Herausforderung darin, komplexe Medieninputs zu verfolgen, mikrobiologische Tests für alle Materialien zu dokumentieren und die kritischen Grenzwerte der Lebensmittelsicherheit einzuhalten - ohne die strengeren Sterilitätsanforderungen der Pharmaindustrie zu erfüllen.
Kontaminations- und Nichtkonformitätstrends
| Merkmal | Biopharmazeutische Produktion | Kultiviertes Fleischherstellung |
|---|---|---|
| Primäre Regulierung | 21 CFR Teile 211 und 600 [2] | FDA/USDA Gemeinsamer Rahmen (HACCP-basiert) |
| Kontaminationsfokus | Klinische Sterilität, aseptische Bekleidung, molekulare Reinheit [2] | Lebensmittelsicherheit, Erregerkontrolle, Rückverfolgbarkeit der Medieneingaben |
| Überprüfungsdauer | 10–15 Tage für Biologika [2] | Unter 5 Tage (Geschwindigkeit der Lebensmittelversorgungskette) |
| Handhabung von Nichtkonformitäten | Formale CAPA im Zusammenhang mit klinischen Sicherheitsauswirkungen [2] | HACCP-basierte Korrekturmaßnahmen konzentrieren sich auf Lebensmittelverderb/Sicherheit |
| Datenintegritätsstandard | ALCOA+ und 21 CFR Teil 11 [1] | Allgemeine GMP- und Lebensmittelsicherheits-Dokumentationsstandards |
Während die menschlichen Fehlerquoten in beiden Branchen ähnlich sind - etwa 50 % der Chargenproblems resultieren aus menschlichen Fehlern [2] - sind die Risiken unterschiedlich.In der Biopharma kann bereits eine einzige nicht dokumentierte Abweichung schwerwiegende Auswirkungen auf die Patientensicherheit haben. Bei kultiviertem Fleisch beziehen sich Kontaminationsrisiken mehr auf lebensmittelbedingte Krankheitserreger und Verderb, die ganze Produktionsläufe beeinträchtigen können.
Fazit
Chargenprotokolle dienen als offizielles Logbuch für jeden Produktionslauf von kultiviertem Fleisch - wenn ein Schritt nicht aufgezeichnet wird, betrachten die Regulierungsbehörden ihn als nicht durchgeführt [6][3]. Dies unterstreicht die Bedeutung präziser Dokumentation und strenger Qualitätskontrolle.
FDA-Inspektionen betonen, dass die Datenintegrität mit den ALCOA+-Prinzipien übereinstimmen muss [1]. Qualitätssicherungsteams müssen alle Produktionsunterlagen überprüfen und genehmigen, bevor eine Charge freigegeben werden kann [2][17], und alle Abweichungen müssen umgehend mit einer dokumentierten Ursachenanalyse untersucht werden [2][5]. Während menschliches Versagen für 50 % der Probleme mit Chargenprotokollen verantwortlich ist, können zweistufige Überprüfungen und strukturierte CAPA-Prozesse (Corrective and Preventive Action) dazu beitragen, diese Risiken zu verringern [2][5].
"Es ist nicht die Komplexität des Prozesses, die zu Beanstandungen führt - es ist die Inkonsistenz, Unvollständigkeit und schlechte Aufsicht." - GXP-Audits & Beratungsdienste [5]
Um diese Herausforderungen zu bewältigen, sollten Produzenten von kultiviertem Fleisch den Fokus auf unabhängige Audits, rigorose Tests von lebensmittelsicheren Zutaten auf mikrobiologische Kontamination und die Sicherstellung der Dokumentation gemäß HACCP- und GCCP-Standards legen. Die Implementierung von elektronischen Chargenaufzeichnungssystemen, validiert gemäß 21 CFR Part 11 [1], kann Fehler erheblich minimieren und den Überprüfungsprozess beschleunigen.
Das regulatorische Umfeld erfordert Präzision, ist jedoch navigierbar.Durch das Lernen aus den Fehlern der Biopharma - wie fehlende Unterschriften bei Qinhuangdao Zizhu Pharmaceutical [17], unzureichende doppelte Verifizierung bei Terumo Corp [18] , und unzureichende Abweichungsdokumentation bei Torrent Pharmaceuticals [18] - können Unternehmen für kultiviertes Fleisch von Anfang an konforme Systeme etablieren. Die Integration dieser Lektionen ermöglicht proaktive Compliance und gleichbleibende Qualität. Sichere Aufbewahrung von Aufzeichnungen, rechtzeitige Berichterstattung über Abweichungen und die Durchführung realistischer Probeaudits werden sicherstellen, dass Chargenaufzeichnungen jederzeit prüfbereit sind und Produktionsläufe vollständig nachvollziehbar bleiben.
Für weitere Ressourcen und Expertenrat zur Aufrechterhaltung hoher Produktionsstandards in der Herstellung von kultiviertem Fleisch besuchen Sie
FAQs
Was sollte ein Chargenprotokoll für kultiviertes Fleisch enthalten?
Ein Chargenprotokoll für kultiviertes Fleisch dient als umfassendes Protokoll des gesamten Herstellungsprozesses. Es muss detaillierte Verarbeitungsanweisungen, schrittweise Ausführungsaufzeichnungen, und alle Abweichungen enthalten, die während der Produktion auftreten. Zusätzlich sollte es In-Prozess-Tests und Freigabetests dokumentieren, um zu bestätigen, dass das Produkt Sicherheits-, Qualitäts- und Regulierungsstandards erfüllt.
Wie können wir die Sterilität anhand von Chargenprotokollen nachweisen?
Der Nachweis der Sterilität durch Chargenprotokolle beinhaltet eine gründliche Prüfung der dokumentierten Sterilisationsverfahren, Testergebnisse und Medienqualitätskontrollberichte, um sicherzustellen, dass sie den behördlichen Anforderungen entsprechen.Es ist entscheidend, Abweichungen oder fehlgeschlagene Tests durch detaillierte Untersuchungen und CAPAs (Korrektur- und Vorbeugemaßnahmen) zu adressieren. Dieser Prozess stellt sicher, dass jeder Schritt eingehalten wurde und alle Probleme ordnungsgemäß gelöst wurden, um die Sterilitätsstandards aufrechtzuerhalten.
Wann sind elektronische Chargenprotokolle (Teil 11) erforderlich?
Elektronische Chargenprotokolle sind unter Teil 11 unerlässlich, wenn elektronische Systeme verwendet werden, um Abweichungen in Chargenprotokollen zu dokumentieren, zu untersuchen und zu rechtfertigen. Sie spielen eine entscheidende Rolle bei der Sicherstellung der Einhaltung von 21 CFR Part 211.192 , Schutz der Datenintegrität, Einhaltung der Untersuchungsfristen und Sicherstellung einer effektiven Managementaufsicht.










