

バッチ記録は、コンプライアンスと製品の安全性にとって重要です。 これらは生産の各ステップを記録し、規制基準が満たされていることを保証します。培養肉の生産者にとって、 無菌性の維持と詳細な記録は譲れない要件です。 FDAの検査では、データの欠如、不完全なレビュー、劣悪な是正措置などの問題がしばしば指摘され、警告や中断につながる可能性があります。
重要なポイント:
- バッチ記録: 2種類 - マスターバッチ記録 (MBR)(「レシピ」)とバッチ生産記録 (BPR)(「実行」)。
- 一般的な問題: 人為的なエラー(問題の50%)、 プロセス中のチェックの欠如、不完全なレビュー、劣悪なCAPA(是正および予防措置)システム。
- FDA基準: ALCOA+原則(帰属可能、判読可能、同時性、オリジナル、正確、完全、一貫性、持続可能、利用可能)への準拠が必須です。
- ソリューション: 独立した監査、電子バッチ記録、厳格なサプライヤー検証は、エラーを最小限に抑え、コンプライアンスを向上させることができます。
UPSIDE Foodsのような培養肉企業は、詳細な文書化、入力のトレーサビリティ、迅速な是正措置を確保することで基準を設定しています。これらの実践から学ぶことで、生産者は規制上の落とし穴を避け、高品質の基準を維持することができます。
ライフサイエンスにおけるFDAコンプライアンスのための文書化と記録保持の包括的ガイド
sbb-itb-ffee270
バッチ記録文書における一般的な問題
FDAの検査報告書は一貫して繰り返される問題を強調しています: 生産記録のレビューの逸脱は、規制当局によって指摘されるGMPの欠陥の中で上位にランクされています[7]. 培養肉の生産者にとって、これらの欠点は単なる管理上のミスを超えて、持続的な無菌状態を示す能力を危険にさらします。これらの問題は、以下の例に示されるように、さまざまな形で現れます。
不完全なレビューと不適合
一般的な問題の一つは、品質管理部門がバッチ記録を徹底的にレビューしないことです。リリースプロセスの重要な部分である代わりに、レビューはしばしば反応的に行われ、製品の問題がすでに発生した後にのみ行われます[7]. このアプローチは、生産記録に大きなギャップを残します。
例えば、Davis City Pharmacy は、成分の量、操作手順、担当者のイニシャルなどの重要な詳細が欠けているため、FDA 483の指摘を受けました。同様に、CAPS は、重要なエントリーに必要な署名とレビュアーの確認が欠けているとして指摘されました[3] . これらの不備は孤立した事例ではありません。研究によれば、約52%の文書違反は、強固なバイオプロセス管理システムが欠如している場合に悪化します[3] .
"プロセスの複雑さが指摘を引き起こすのではなく、一貫性の欠如、不完全さ、そして不十分な監視が原因です。" - GXP監査&コンサルティングサービス[5]
プロセス中のチェック記録の欠如
もう一つの頻繁な欠陥は、プロセス中のチェックに関する適切な文書の欠如です。これらの記録は、無菌操作の重要な管理点で特に重要です。例えば、ネフロン無菌調剤センターは、バッチ記録における重要なガウン着用手順や無菌手順の文書化を怠ったことで指摘を受けました[3] . 培養肉の生産者にとって、無菌性が最重要であるため、そのような省略は汚染管理措置.
の遵守を確認することを不可能にします。Amphastarは、予期しない収量の変動や出力の不一致を調査または文書化しなかったためにも指摘されました[3]. そのような見落としのリスクは明白です。あるケースでは、2024/2025年に特定されていない製薬施設が、完成したバッチ記録を開放棚や机の上に保管していることが判明しました。FDAの調査官は、1つの記録から7ページを含む欠落ページや、別の記録から「合成溶液」セクション全体が欠如していることを発見しました[6].
CAPAとサプライヤーGMP検証の失敗
文書エラーを超えて、効果的な是正プロセスとサプライヤー検証の欠如は、バッチ記録の信頼性をさらに損ないます。生産の逸脱が対応する是正および予防措置(CAPA)報告書なしに発生すると、バッチ記録の整合性が損なわれます[7]. 例えば、Eugia Pharma Specialities Limited, は2024年1月22日から2月2日までの間に検査を受け、不備を適切にレビューしなかったためにFDA 483を受け取りました。彼らの非効率なCAPAシステムと不完全な調査は、生産問題の繰り返しを引き起こし、調査とCAPA手順の完全な見直しを余儀なくされました[9].
同様に、2023年9月26日から10月25日までの検査中に、Stokes Healthcare Inc.は不十分な不一致管理を示しました。同社は影響を受けたすべてのバッチに調査を拡大することができず、分析の完了を遅らせました[9].
「CAPAや逸脱報告書のない不一致?それはコンプライアンスの失敗です。" - GXP監査 & コンサルティングサービス [5]
サプライヤーの検証問題は、さらに複雑さを増します。Empower Clinic Services LLCは、2022年7月18日から8月5日までの検査で、不十分なサプライヤーの資格と不適切な調査プロセスを含む不十分な品質管理手順で指摘されました[9] . 成長媒体、細胞株、その他の重要な投入物に依存する培養肉生産者にとって、サプライヤーのGMPコンプライアンスを確保することは、バッチ記録の整合性を維持するために重要です。
バッチ記録に関するFDAの要件
バッチ記録に関するFDAの規則は、21 CFR Part 117, に基づいており、食品安全の基準を設定しています。培養肉に関しては、細胞培養段階での無菌状態の維持が重要であり、文書はしばしばPart 111またはPart 211 , に加えてPart 117[10][14]. の厳しい基準を満たす必要があります。これは、培養肉の生産の安全性と有効性を確保するために、正確な文書がいかに重要であるかを強調しています。
バッチ記録の基本基準
各バッチには2つの重要な文書が必要です:
BPRには、バッチまたはロット番号、機器の詳細、清掃日、部品識別子、正確な測定値、実際の収量と理論的収量の比較などの詳細を含める必要があります[10][14].
「バッチ生産記録は、適切なマスタ製造記録に正確に従わなければならず、バッチの生産における各ステップを実行しなければなりません。」 – 21 CFR 111.255 [12]
すべての重要なステップは、実行者と確認者のイニシャルを記載して直ちに記録されなければなりません[10][11]. FDAはALCOA(+)原則の遵守を要求しており、記録は帰属可能、判読可能、同時性、オリジナル、正確である必要があります - さらに完全、一貫、持続、利用可能 [1] .
マスターマニュファクチャリングレコードからの逸脱がある場合、徹底的に調査する必要があります。これには、問題の記録、根本原因分析の実施、および是正および予防措置 (CAPA)プランの実施が含まれます[8] [1]. 逸脱の初期評価は、検出から24〜48時間以内に記録する必要があります[8]. 電子システムを使用する施設では、21 CFR Part 11への準拠が必須です。これには、検証済みの電子署名と安全でタイムスタンプ付きの監査証跡が含まれます[8] [1].
記録の保持とレビュー手順
適切な記録の保持とレビューのプロセスは、コンプライアンスを維持し、製品の安全性を確保するために重要です。無菌生産、例えば培養肉の生産では、バッチ記録のすべての詳細が綿密にレビューされなければなりません。品質管理(QC)チームは、すべてのバッチ記録をレビューし、結果を監視し、データをテストして、バッチが流通のために承認される前に責任を負います[10] [13].
「すべての医薬品製造および管理記録は、バッチがリリースまたは流通される前に品質管理部門によってレビューおよび承認されなければなりません。」– 21 CFR 211.192 [2]
製造業者はしばしば、生産から30日以内に95%のバッチレビューを完了することを目指しています[2]. しかし、培養肉に関わるより複雑な無菌プロセスでは、レビューには通常 7~10日, かかり、高性能な施設では7日未満の時間を達成しています[2]. 電子バッチ記録システムは、培養肉生産システム, に統合されたものなど、これらのレビューを大幅に迅速化できます - 紙ベースの方法と比較して時間を半分に短縮 - ただし、Part 11の要件を満たし、データの完全性を維持するために検証されている限りです[1].
FDA承認の培養肉企業が成功した理由
FDA承認の培養肉企業は、文書化の課題に対応し、厳格な安全基準を満たす実践を採用することで、高い基準を設定しました。
UPSIDE Foodsが2022年11月にFDAの事前市場相談を通過した最初の培養肉企業となったとき、彼らは業界のモデルを確立しました。FDAは、細胞株の確立、細胞バンク、製造管理、すべての構成要素と入力を含む生産プロセスを徹底的にレビューした後、「これ以上の質問はない」というレターを発行しました。この成果は、FDAの厳しい要件を満たすための詳細な文書化の重要性を強調しました。 無菌性とコンプライアンス基準の達成 UPSIDE Foodsの際立った成果は、入力のトレーサビリティに対する徹底的なアプローチでした。すべての生産要素が慎重に文書化され、初期の細胞株から最終製品までの明確な責任の連鎖が確保されました。この透明性のレベルにより、FDAのレビュアーは製造プロセスのすべてのステップを追跡し、すべての安全基準が一貫して満たされていることを確認できました。
"FDAの企業との事前相談には、企業の生産プロセスとそのプロセスで作られた培養細胞材料の評価が含まれていました。これには、一次および不死化細胞株と細胞バンクの確立、製造管理、すべての構成要素と投入物が含まれます。" – U.S. 食品医薬品局 [16]
他の成功した企業は、詳細な無菌プロセス文書化を実施することで追随しました。これには、ガウン着用手順や無菌操作などの重要なステップが含まれていました [3]. 以前の文書化の失敗とは異なり、これらの企業は、オペレーターのチェック、生産監督、品質部門のレビューを含む階層化されたレビューシステムを採用し、バッチリリース前に潜在的なエラーを検出しました [15]. 電子バッチ記録システムも重要な役割を果たし、各段階での必須の署名を強制し、21 CFR Part 11の要件に従って不変の監査証跡を維持しました [3][2].
これらの厳格な慣行は、企業が逸脱や失敗をどのように処理するかにも自然に拡張されました。
バッチ失敗のためのCAPAプロセス
バッチが仕様を満たさなかった場合、FDA承認企業は迅速かつ体系的に行動しました。彼らの是正および予防措置(CAPA)プロセスには、正式な根本原因分析、影響評価、および明確に文書化された是正措置が含まれていました[3]. いかなる逸脱も統合された品質保証フレームワーク内で管理され、生産が続行される前にすべての問題が徹底的に調査され、正当化され、文書化されることを保証しました[2].
今後、データの完全性は2024年から2025年にかけてFDAの執行行動の主要な焦点となる予定です[1].
バッチ記録の実践を改善する方法
バッチ記録の実践を強化するには、FDAの検査でよく指摘される一般的な失敗に対処するための正確な文書化が必要です。ここでは、主要な課題に取り組むためのいくつかの戦略を紹介します。
独立したバッチ記録監査を実施する
定期的な第三者監査は、内部レビューでは見落とされがちな問題を発見することができます。まず、LIMS(ラボ情報管理システム)、MES(製造実行システム)、ERP(企業資源計画)などの重要なシステムに焦点を当てます。リリーステスト記録、安定性データ、バッチ生産記録など、規制上の影響が大きい文書を優先します。
効果的な方法の一つは、サンプル回収テストです。最近のバッチをランダムに選択し、その生産および実験室の履歴を再構築します。これにより、欠落しているデータ、不完全な署名、または規制上の指摘につながる可能性のある文書のギャップを特定するのに役立ちます。システム生成の監査証跡を手動入力と照合し、不正な変更や削除を特定します。
過去1年間の規格外(OOS)および傾向外(OOT)レポートをすべて確認します。根本原因分析が徹底されているか、是正および予防措置(CAPA)が適切に実施されているかを評価します。文書の問題がFDA警告書の21%を占めている一方で、人為的ミスが製薬製造におけるバッチ記録の問題の50%を占めていることは注目に値します。[2].
「指摘を引き起こすのはプロセスの複雑さではなく、一貫性の欠如、不完全さ、そして不十分な監督です。" – GXP監査 & コンサルティングサービス [5]
定期的な模擬レビューを通じて規制検査をシミュレートします。この実践により、チームは実際の監査の前に不整合や潜在的なデータの完全性の問題を認識するのに役立ちます。すべての記録がALCOA+の原則に従っていることを確認してください:帰属可能、判読可能、同時性、オリジナル、正確、完全、一貫性、持続可能、利用可能。
文書の完全性が確立されたら、すべての生産入力の品質を確認することに集中してください。
すべての入力の微生物汚染をテストする
すべての入力に対する独立した無菌性および効力試験は不可欠です - サプライヤーの分析証明書(CoA)にのみ依存しないでください。これは特に培養肉の生産者にとって重要であり、汚染がバッチ全体を危険にさらす可能性があります。
例えば、2013年2月に、 Central Admixture Pharmacy Servicesは、無菌製品のバッチリリース時の微生物管理が不十分であるとしてFDAからの指摘を受けました。同社は、標準作業手順書(SOPs)に詳細な微生物管理手順を導入しなければなりませんでした。[4].
プロセス中の微生物チェックポイントは、最終製品試験への過度の依存を防ぐことができます。これらのチェックポイントをバッチリリースSOPsに組み込み、厳格な同時記録を維持してください。すべての試験結果と製造工程をその場で記録し、バックデートや遅延入力を避けることで、FDAからの指摘を防ぎます。
供給業者ファイルを包括的に保管し、CoA、監査報告書、品質契約、入荷材料に関連する逸脱の履歴を含めてください。
バッチ記録の実践を強化するには、HACCPやGCCPのような確立された基準にプロセスを合わせることがさらに求められます。
HACCPおよびGCCP基準に合わせた記録
バッチ記録にハザード分析重要管理点(HACCP)の原則を組み込むことで、重要なプロセス変数が生産全体で監視および記録されることを保証します。これには、最終段階のテストにのみ依存するのではなく、プロセス中の微生物検査のチェックポイントを確立することが含まれます。
培養肉の生産者にとって、良好な細胞培養実践(GCCP)基準の遵守も同様に重要です。バッチ記録には、無菌操作、ガウン着用手順、およびバッチリリース基準に関連する環境モニタリングの詳細を含める必要があります[3][4]. これらの手順は、コンプライアンスを維持し、製品の安全性を確保するのに役立ちます。
業界データによると、適切なバッチ製造ソフトウェアが導入されていない場合、文書違反の52%がエスカレートします[3][4]. 一例として、2023年2月にNephron Sterile Compounding Centreは、バッチリリース前に重要なプロセス変数を検証するための管理手順が欠如しているため、FDAの観察を受けました[4]. これは、認識された基準に沿った積極的な文書化の必要性を強調しています。
電子バッチ記録(EBR)への移行は、リアルタイムのデータ収集と自動化されたワークフローを通じて、文書エラーを最大50%削減することができます[2]. これらのシステムは、バッチが進む前に微生物試験結果の欠如やレビューの不完全さを警告し、人為的なエラーを最小限に抑えます。
「FDAは記録がALCOA(+)であることを期待しています:帰属可能、判読可能、同時性、オリジナル、正確 - さらに完全、一貫性、持続性、利用可能であること。" – Atlas Compliance [1]
バッチ記録における説明のつかない不一致や逸脱は、正式な調査およびCAPAシステムにリンクされるべきです。電子的な微生物試験データの整合性を保護するために、書き込みおよび削除の権限を制限してください。競争力のある製造業者は、生産完了後30日以内に95%のバッチをレビューしリリースすることを目指しています[2].
これらの行動は、引用のリスクを減らすだけでなく、最近のFDA検査で強調された厳格な文書化基準にも合致しています。
バイオファーマ vs 培養肉: バッチ記録の違い
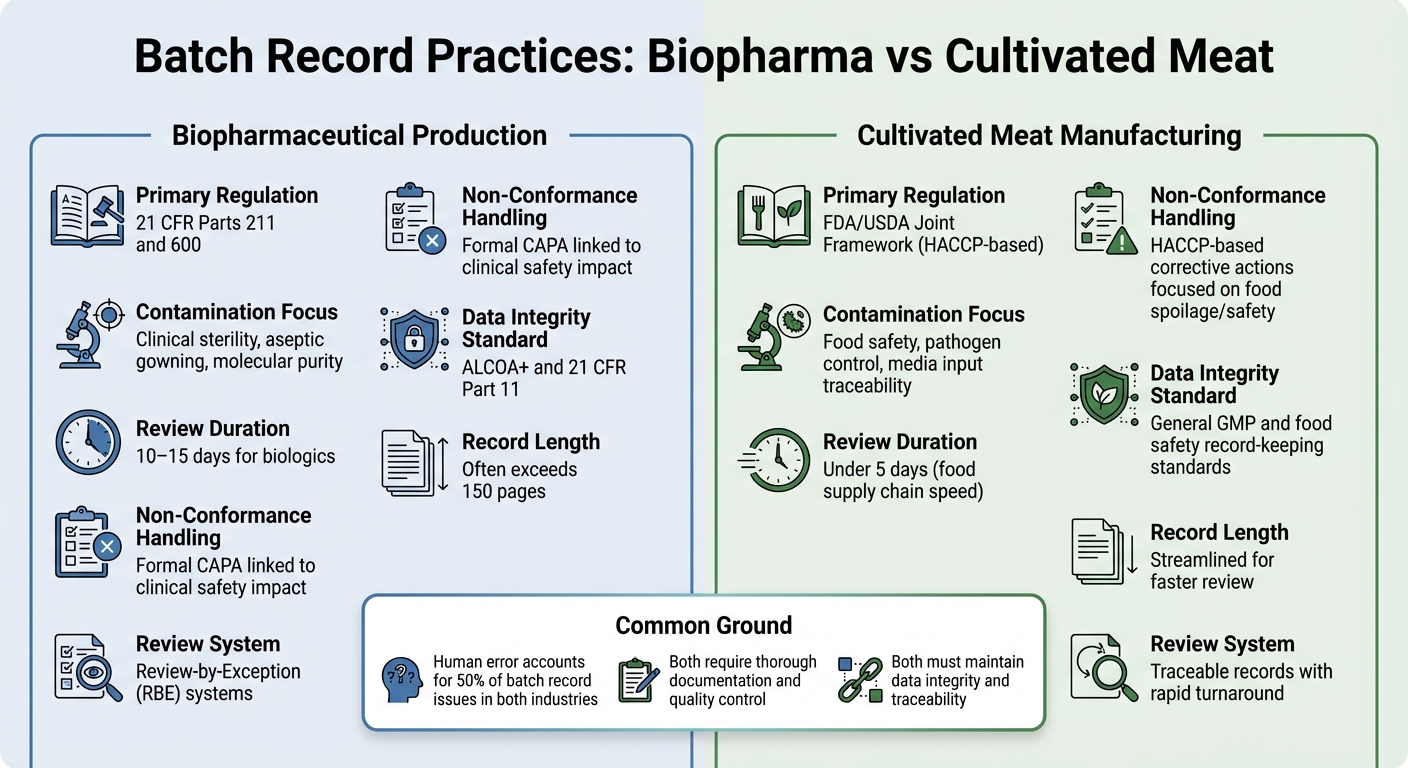
バイオ医薬品 vs 培養肉 バッチ記録要件の比較
バイオ医薬品の生産と培養肉の製造におけるバッチ記録の実践の違いを見ていくと、これらの業界における規制上の要求がどのように文書化の優先順位を形作っているかがより明確になります。
両セクターとも徹底した文書化が求められますが、規制の枠組みや管理目標は大きく異なります。バイオファーマでは、バッチ記録は21 CFR パート 211 および 600, の下で厳しく規制されており、品質管理部門がバッチをリリースする前にすべての生産および管理記録をレビューし、承認する必要があります[2]. 一方、培養肉の生産者は通常、HACCPおよびGCCP基準に従います。これらは、注射用バイオ医薬品に要求される臨床グレードの無菌性ではなく、食品の安全性と病原体の制御に重点を置いています。
バイオファーマのバッチ記録はしばしば膨大で、150ページを超えることもあり、レビューには10~15日かかることがあります。これを効率化するために、多くのバイオファーマ企業は例外によるレビュー (RBE)システムを使用しており、主要な逸脱をコンパクトなレポートにまとめています。一方、培養肉の生産者は、食品供給チェーンの迅速なペースを反映して、5日以内にレビューできる追跡可能な記録を目指しています。 [2].
これらの記録の内容は、優先事項の違いも浮き彫りにしています。バイオファーマの検査は、ガウンの手順や環境管理などの無菌処理の詳細に焦点を当てることが多いです。対照的に、培養肉の記録は、食品の安全性を確保するために培地の投入物や微生物学的試験を強調する必要があります。培養肉においては、複雑な培地の入力を追跡し、すべての材料の微生物学的試験を記録し、食品安全の重要限界を満たすことが課題です - 医薬品の厳しい無菌要件に従うことなく。
汚染と不適合の傾向
| 特徴 | バイオ医薬品製造 | 培養肉製造 |
|---|---|---|
| 主要規制 | 21 CFR パート 211 および 600[2] | FDA/USDA 共同フレームワーク(HACCPベース) |
| 汚染の焦点 | 臨床的無菌性、無菌ガウン、分子純度[2] | 食品安全、病原体管理、メディア入力のトレーサビリティ |
| レビュー期間 | バイオ医薬品の場合は10~15日[2] | 5日以内(食品供給チェーンの速度) |
| 不適合の処理 | 臨床安全性への影響に関連する正式なCAPA [2] | 食品の腐敗/安全性に焦点を当てたHACCPベースの是正措置 |
| データインテグリティ基準 | ALCOA+および21 CFR Part 11 [1] | 一般的なGMPおよび食品安全記録保持基準 |
人的エラー率は両業界で同様であるが - バッチ記録の問題の約50%は人間のミスに起因する[2] - 賭け金は異なる。バイオファーマでは、たった一つの未記録の逸脱でも患者の安全に重大な影響を及ぼす可能性があります。培養肉の場合、汚染リスクは食中毒病原体や腐敗に関するものであり、これが生産全体に影響を及ぼす可能性があります。
結論
バッチ記録は、培養肉の各生産ランの公式ログとして機能します - ステップが記録されていない場合、規制当局はそれが実行されていないと見なします [6][3]. これは、正確な文書化と厳格な品質管理の重要性を強調しています。
FDAの検査は、データの完全性がALCOA+の原則に沿っている必要があることを強調しています [1]. 品質管理チームは、バッチがリリースされる前にすべての生産記録をレビューし、承認する必要があります[2] [17], また、逸脱が発生した場合は、文書化された根本原因分析を伴う迅速な調査が必要です[2][5]. バッチ記録の問題の50%は人為的ミスによるものですが、二重レベルのレビューと構造化されたCAPA(是正および予防措置)プロセスにより、これらのリスクを軽減することができます[2][5].
「引用を引き起こすのはプロセスの複雑さではなく、一貫性の欠如、不完全さ、そして不十分な監視です。" - GXP監査 & コンサルティングサービス [5]
これらの課題を克服するために、培養肉の生産者は独立した監査、微生物汚染に対する食品安全な成分の厳格なテスト、およびHACCPとGCCP基準に準拠した文書化に注力する必要があります。21 CFR Part 11の下で検証された電子バッチ記録システムを実装することにより、エラーを大幅に最小化し、レビューのプロセスを迅速化できます。
規制環境は精密さを要求しますが、ナビゲート可能です。バイオファーマのエラーから学ぶことで、培養肉企業は最初からコンプライアンスシステムを確立することができます。これらの教訓を取り入れることで、積極的なコンプライアンスと一貫した品質を実現できます。記録の安全な保持、逸脱報告のタイムリーな実施、現実的な模擬監査の実施により、バッチ記録が検査準備完了状態を維持し、生産ランが完全に追跡可能であることを保証します。
培養肉製造における高い生産基準を維持するためのリソースと専門家のガイダンスについては、
よくある質問
培養肉のバッチ記録には何を含めるべきですか?
培養肉のバッチ記録は、製造プロセス全体の包括的なログとして機能します。これには、詳細な処理手順, ステップバイステップの実行記録, および生産中に発生する 逸脱の記録が含まれている必要があります。さらに、プロセス中のテストおよび出荷前テストを記録し、製品が安全性、品質、および規制基準を満たしていることを確認する必要があります。
バッチ記録を使用して無菌性を証明するにはどうすればよいですか?
バッチ記録を通じて無菌性を証明するには、文書化された滅菌手順、テスト結果、およびメディア品質管理レポートを徹底的に検査し、それらが規制要件を満たしていることを確認する必要があります。逸脱や試験の失敗は、詳細な調査とCAPA(是正および予防措置)を通じて対処することが重要です。このプロセスは、すべてのステップが遵守され、問題が適切に解決されて無菌基準が維持されることを保証します。
電子バッチ記録(Part 11)はいつ必要ですか?
電子バッチ記録は、電子システムがバッチ記録の逸脱を文書化、調査、正当化するために使用される場合、Part 11の下で不可欠です。これらは、21 CFR Part 211.192, の遵守を確保し、データの完全性を保護し、調査のタイムラインを満たし、効果的な管理監督を確保する上で重要な役割を果たします。









