

बैच रिकॉर्ड अनुपालन और उत्पाद सुरक्षा के लिए महत्वपूर्ण हैं। वे उत्पादन के हर चरण का दस्तावेजीकरण करते हैं, यह सुनिश्चित करते हुए कि नियामक मानकों का पालन किया जा रहा है। कृत्रिम मांस उत्पादकों के लिए, स्वच्छता बनाए रखना और विस्तृत रिकॉर्ड रखना अनिवार्य है। एफडीए निरीक्षण अक्सर डेटा की कमी, अधूरी समीक्षाओं, और खराब सुधारात्मक कार्रवाइयों जैसे मुद्दों को उजागर करते हैं, जो चेतावनियों या व्यवधानों का कारण बन सकते हैं।
मुख्य निष्कर्ष:
- बैच रिकॉर्ड: दो प्रकार - मास्टर बैच रिकॉर्ड (एमबीआर) ("रेसिपी") और बैच उत्पादन रिकॉर्ड (बीपीआर) ("कार्यान्वयन")।
- सामान्य समस्याएं: मानव त्रुटियाँ (50% समस्याएं), प्रक्रिया में जाँच, की कमी, अधूरी समीक्षाएं, और खराब CAPA (सुधारात्मक और निवारक कार्रवाई) प्रणाली।
- एफडीए मानक: ALCOA+ सिद्धांतों (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available) का पालन अनिवार्य है।
- समाधान: स्वतंत्र ऑडिट, इलेक्ट्रॉनिक बैच रिकॉर्ड, और कठोर आपूर्तिकर्ता सत्यापन त्रुटियों को कम कर सकते हैं और अनुपालन में सुधार कर सकते हैं।
संवर्धित मांस कंपनियाँ जैसे UPSIDE Foods ने विस्तृत दस्तावेज़ीकरण, इनपुट ट्रेसबिलिटी, और त्वरित सुधारात्मक उपाय सुनिश्चित करके एक मानक स्थापित किया है। इन प्रथाओं से सीखकर, उत्पादक नियामक समस्याओं से बच सकते हैं और उच्च गुणवत्ता मानकों को बनाए रख सकते हैं।
जीवन विज्ञान में FDA अनुपालन के लिए दस्तावेज़ीकरण और रिकॉर्ड-कीपिंग के लिए व्यापक गाइड
sbb-itb-ffee270
बैच रिकॉर्ड दस्तावेज़ीकरण में सामान्य समस्याएँ
FDA निरीक्षण रिपोर्ट लगातार एक आवर्ती मुद्दे को उजागर करती हैं: उत्पादन रिकॉर्ड समीक्षा विचलन शीर्ष GMP कमियों में से एक है जिसे नियामकों द्वारा उद्धृत किया गया है [7]. संवर्धित मांस उत्पादकों के लिए, ये कमियाँ केवल प्रशासनिक त्रुटियों तक सीमित नहीं हैं - वे निरंतर स्वच्छ परिस्थितियों को प्रदर्शित करने की क्षमता को खतरे में डालती हैं। ये समस्याएँ कई रूपों में प्रकट होती हैं, जैसा कि नीचे दिए गए उदाहरणों में दर्शाया गया है।
अपूर्ण समीक्षाएँ और गैर-अनुरूपताएँ
एक सामान्य समस्या यह है कि गुणवत्ता नियंत्रण इकाइयाँ बैच रिकॉर्ड्स की पूरी तरह से समीक्षा करने में विफल रहती हैं। रिलीज प्रक्रिया का अभिन्न हिस्सा होने के बजाय, समीक्षाएँ अक्सर प्रतिक्रियात्मक रूप से होती हैं - केवल तब जब कोई उत्पाद समस्या पहले से ही उभर चुकी होती है [7]. यह दृष्टिकोण उत्पादन रिकॉर्ड्स में महत्वपूर्ण अंतराल छोड़ देता है।
उदाहरण के लिए, डेविस सिटी फार्मेसी को बैच रिकॉर्ड्स में घटक मात्राएँ, संचालन चरण, और कर्मियों के हस्ताक्षर जैसी महत्वपूर्ण जानकारी के अभाव के कारण एफडीए 483 अवलोकन प्राप्त हुआ। इसी तरह, CAPS को प्रमुख प्रविष्टियों में आवश्यक हस्ताक्षर और समीक्षक सत्यापन की कमी के लिए उद्धृत किया गया [3]. ये चूकें अलग-थलग घटनाएँ नहीं हैं; अध्ययन दिखाते हैं कि लगभग 52% दस्तावेज़ उल्लंघन बढ़ जाते हैं जब मजबूत बायोप्रोसेस प्रबंधन प्रणाली अनुपस्थित होती हैं [3].
"यह प्रक्रिया की जटिलता नहीं है जो उद्धरणों को प्रेरित करती है - यह असंगति, अपूर्णता, और खराब निगरानी है।" - जीएक्सपी ऑडिटिंग & परामर्श सेवाएँ [5]
प्रक्रिया में जाँच रिकॉर्ड गायब
एक और सामान्य कमी प्रक्रिया में जाँच के लिए उचित दस्तावेज़ की अनुपस्थिति है। ये रिकॉर्ड विशेष रूप से एसेप्टिक संचालन में महत्वपूर्ण नियंत्रण बिंदुओं पर महत्वपूर्ण होते हैं। उदाहरण के लिए, नेफ्रॉन स्टेराइल कंपाउंडिंग सेंटर को उनके बैच रिकॉर्ड में आवश्यक गाउनिंग चरणों और एसेप्टिक प्रक्रियाओं को दस्तावेज़ करने में विफल रहने के लिए उद्धरणों का सामना करना पड़ा [3]. संवर्धित मांस उत्पादकों के लिए, जहाँ स्वच्छता अत्यंत महत्वपूर्ण है, ऐसी चूकें संक्रमण नियंत्रण उपायों.
के अनुपालन की पुष्टि करना असंभव बना देती हैं।एम्फास्टार को भी अप्रत्याशित उपज भिन्नताओं या उत्पादन विसंगतियों की जांच या दस्तावेजीकरण में विफल रहने के लिए चिह्नित किया गया था [3]. ऐसी चूकों के जोखिम स्पष्ट हैं। एक मामले में, 2024/2025 में एक अज्ञात फार्मास्यूटिकल सुविधा को खुले शेल्विंग और डेस्क पर पूर्ण बैच रिकॉर्ड संग्रहीत करते हुए पाया गया। एफडीए जांचकर्ताओं ने गायब पृष्ठों की खोज की, जिसमें एक ही रिकॉर्ड से सात पृष्ठ शामिल थे, और एक अन्य से "संश्लेषण समाधान" अनुभाग पूरी तरह से अनुपस्थित था [6].
CAPA और आपूर्तिकर्ता GMP सत्यापन विफलताएँ
दस्तावेज़ त्रुटियों से परे, प्रभावी सुधारात्मक प्रक्रियाओं और आपूर्तिकर्ता सत्यापन की अनुपस्थिति बैच रिकॉर्ड की विश्वसनीयता को और कमजोर करती है।जब उत्पादन विचलन बिना संबंधित सुधारात्मक और निवारक कार्रवाई (CAPA) रिपोर्ट के होते हैं, तो बैच रिकॉर्ड की अखंडता से समझौता होता है [7]. उदाहरण के लिए, Eugia Pharma Specialities Limited, 22 जनवरी से 2 फरवरी 2024 के बीच निरीक्षण किया गया, और उन्हें असंगतियों की पर्याप्त समीक्षा करने में विफल रहने के लिए FDA 483 प्राप्त हुआ। उनकी अप्रभावी CAPA प्रणाली और अधूरी जांचों ने बार-बार उत्पादन समस्याओं को जन्म दिया, जिससे उनकी जांच और CAPA प्रक्रियाओं का पूर्ण पुनर्गठन करना पड़ा [9].
इसी तरह, 26 सितंबर से 25 अक्टूबर 2023 के बीच एक निरीक्षण के दौरान, Stokes Healthcare Inc. ने खराब असंगति प्रबंधन का प्रदर्शन किया। कंपनी सभी प्रभावित बैचों तक जांच का विस्तार करने में विफल रही और अपनी विश्लेषणों को पूरा करने में देरी की [9].
"कोई CAPA या विचलन रिपोर्ट के बिना असंगति? यह अनुपालन की विफलता है।" - GXP ऑडिटिंग & परामर्श सेवाएँ [5]
आपूर्तिकर्ता सत्यापन मुद्दे जटिलता की एक और परत जोड़ते हैं। एम्पावर क्लिनिक सर्विसेज एलएलसी को 18 जुलाई से 5 अगस्त 2022 के बीच निरीक्षण के दौरान अपर्याप्त गुणवत्ता नियंत्रण प्रक्रियाओं के लिए उद्धृत किया गया था, जिसमें अपर्याप्त आपूर्तिकर्ता योग्यताएँ और खराब जांच प्रक्रियाएँ शामिल थीं [9]. संवर्धित मांस उत्पादकों के लिए, जो वृद्धि मीडिया, सेल लाइनों और अन्य महत्वपूर्ण इनपुट्स पर निर्भर करते हैं, आपूर्तिकर्ता जीएमपी अनुपालन सुनिश्चित करना बैच रिकॉर्ड्स की अखंडता बनाए रखने के लिए महत्वपूर्ण है।
बैच रिकॉर्ड्स के लिए एफडीए आवश्यकताएँ
एफडीए के बैच रिकॉर्ड्स के नियम 21 सीएफआर पार्ट 117, के चारों ओर घूमते हैं जो खाद्य सुरक्षा के लिए आधार रेखा निर्धारित करता है।जब बात कृत्रिम मांस की आती है, जहाँ सेल कल्चर चरण के दौरान स्वच्छता बनाए रखना महत्वपूर्ण होता है, दस्तावेज़ीकरण को अक्सर भाग 111 या भाग 211, के सख्त मानकों को पूरा करना पड़ता है, इसके अलावा भाग 117 [10][14]. यह दर्शाता है कि कृत्रिम मांस उत्पादन की सुरक्षा और प्रभावशीलता सुनिश्चित करने के लिए सटीक दस्तावेज़ीकरण कितना आवश्यक है।
बैच रिकॉर्ड्स के लिए मुख्य मानक
प्रत्येक बैच के लिए दो मुख्य दस्तावेज़ आवश्यक होते हैं:
- मास्टर बैच रिकॉर्ड (MBR): उत्पादन प्रक्रिया को दर्शाने वाला स्वीकृत टेम्पलेट।
- बैच उत्पादन रिकॉर्ड (BPR): उत्पादन रन के दौरान वास्तव में क्या होता है इसका विस्तृत रिकॉर्ड [12][2].
बीपीआर में बैच या लॉट नंबर, उपकरण विवरण, सफाई की तारीखें, घटक पहचानकर्ता, सटीक माप, और वास्तविक बनाम सैद्धांतिक उपज की तुलना जैसी विशिष्टताएँ शामिल होनी चाहिए [10][14].
"बैच उत्पादन रिकॉर्ड को उपयुक्त मास्टर मैन्युफैक्चरिंग रिकॉर्ड का सही तरीके से पालन करना चाहिए और आपको बैच के उत्पादन के प्रत्येक चरण को पूरा करना चाहिए।" – 21 सीएफआर 111.255 [12]
प्रत्येक महत्वपूर्ण चरण को तुरंत रिकॉर्ड किया जाना चाहिए, जिसमें प्रदर्शनकर्ता और सत्यापनकर्ता के प्रारंभिक हस्ताक्षर नोट किए गए हों [10][11]. एफडीए एएलसीओए(+) सिद्धांतों का पालन करने की आवश्यकता होती है, जिसका अर्थ है कि रिकॉर्ड संबद्ध, पठनीय, समकालीन, मूल, और सटीक - साथ ही पूर्ण, सुसंगत, स्थायी, और उपलब्ध [1].
यदि मास्टर मैन्युफैक्चरिंग रिकॉर्ड से कोई विचलन होता है, तो इसकी पूरी तरह से जांच की जानी चाहिए। इसमें समस्या का दस्तावेजीकरण, मूल कारण विश्लेषण करना, और सुधारात्मक और निवारक कार्रवाई (CAPA) योजना लागू करना शामिल है[8] [1]. विचलनों के प्रारंभिक आकलन का पता चलने के 24-48 घंटों के भीतर लॉग किया जाना चाहिए[8]. इलेक्ट्रॉनिक सिस्टम का उपयोग करने वाली सुविधाओं के लिए, 21 CFR Part 11 का अनुपालन अनिवार्य है। इसमें सत्यापित इलेक्ट्रॉनिक हस्ताक्षर और सुरक्षित, समय-चिह्नित ऑडिट ट्रेल्स शामिल हैं[8] [1].
रिकॉर्ड प्रतिधारण और समीक्षा प्रक्रियाएँ
उचित रिकॉर्ड प्रतिधारण और समीक्षा प्रक्रियाएँ अनुपालन में बने रहने और उत्पाद सुरक्षा सुनिश्चित करने के लिए महत्वपूर्ण हैं।In sterile production, like that of cultivated meat, every detail in the batch records must undergo meticulous review. The Quality Control (QC) team is responsible for reviewing all batch records, monitoring results, and testing data before a batch can be approved for distribution [10] [13].
"सभी दवा उत्पाद उत्पादन और नियंत्रण रिकॉर्ड्स को बैच जारी या वितरित करने से पहले गुणवत्ता नियंत्रण इकाई द्वारा समीक्षा और अनुमोदित किया जाना चाहिए।" – 21 CFR 211.192 [2]
निर्माता अक्सर उत्पादन के 30 दिनों के भीतर [2] . 95% बैच समीक्षाओं को पूरा करने का लक्ष्य रखते हैं। हालांकि, संवर्धित मांस में शामिल अधिक जटिल स्टरल प्रक्रियाओं के लिए, समीक्षाएं आमतौर पर 7–10 दिनों, का समय लेती हैं, उच्च प्रदर्शन करने वाली सुविधाएं 7 दिनों से कम समय में समय प्राप्त करती हैं [2]. इलेक्ट्रॉनिक बैच रिकॉर्ड सिस्टम इन समीक्षाओं को काफी तेजी से कर सकते हैं, जैसे कि संवर्धित मांस उत्पादन प्रणालियों, में एकीकृत - कागज आधारित विधियों की तुलना में समय को आधा कर देते हैं - जब तक कि वे भाग 11 आवश्यकताओं को पूरा करने और डेटा अखंडता बनाए रखने के लिए मान्य हैं [1].
एफडीए-अनुमोदित संवर्धित मांस कंपनियों ने क्या सही किया
एफडीए-अनुमोदित संवर्धित मांस कंपनियों ने दस्तावेज़ीकरण चुनौतियों का समाधान करने और कठोर सुरक्षा मानकों को पूरा करने वाली प्रथाओं को अपनाकर उच्च मानक स्थापित किया है।
जब UPSIDE Foods नवंबर 2022 में एफडीए की पूर्व-बाजार परामर्श पास करने वाली पहली संवर्धित मांस कंपनी बनी, तो उन्होंने उद्योग के लिए एक मॉडल स्थापित किया।FDA ने उनके उत्पादन प्रक्रिया की पूरी समीक्षा करने के बाद "कोई और प्रश्न नहीं" पत्र जारी किया, जिसमें सेल लाइन स्थापना, सेल बैंक, निर्माण नियंत्रण, और सभी घटक और इनपुट शामिल थे [16]. इस उपलब्धि ने FDA की सख्त आवश्यकताओं को पूरा करने में विस्तृत दस्तावेज़ीकरण के महत्व को उजागर किया।
स्वच्छता और अनुपालन मानकों को पूरा करना
UPSIDE Foods की प्रमुख उपलब्धि उनके इनपुट ट्रेसबिलिटी के व्यापक दृष्टिकोण में थी। हर उत्पादन घटक को सावधानीपूर्वक दस्तावेज़ित किया गया, जिससे प्रारंभिक सेल लाइन से अंतिम उत्पाद तक की स्पष्ट जिम्मेदारी श्रृंखला सुनिश्चित हुई [16]. इस स्तर की पारदर्शिता ने FDA समीक्षकों को उत्पादन प्रक्रिया के हर चरण का पता लगाने की अनुमति दी, यह पुष्टि करते हुए कि सभी सुरक्षा मानकों को लगातार पूरा किया गया।
"एफडीए की प्री-मार्केट परामर्श में फर्म की उत्पादन प्रक्रिया और उत्पादन प्रक्रिया द्वारा बनाए गए संवर्धित सेल सामग्री का मूल्यांकन शामिल था, जिसमें प्राथमिक और अमर कोशिका रेखाओं और सेल बैंकों की स्थापना, विनिर्माण नियंत्रण, और सभी घटक और इनपुट शामिल थे।" – U.S. फूड एंड ड्रग एडमिनिस्ट्रेशन [16]
अन्य सफल कंपनियों ने विस्तृत एसेप्टिक प्रक्रिया दस्तावेज़ीकरण को लागू करके इसका अनुसरण किया। इसमें गाउनिंग प्रक्रियाओं और स्टेराइल हैंडलिंग ऑपरेशंस जैसे महत्वपूर्ण कदम शामिल थे [3]. पहले की दस्तावेज़ीकरण विफलताओं के विपरीत, इन कंपनियों ने टियरड समीक्षा प्रणालियों को अपनाया, जिसमें ऑपरेटर चेक, उत्पादन पर्यवेक्षण, और गुणवत्ता इकाई समीक्षाएं शामिल थीं, ताकि बैच रिलीज से पहले संभावित त्रुटियों को पकड़ा जा सके [15]. इलेक्ट्रॉनिक बैच रिकॉर्ड सिस्टम ने भी एक महत्वपूर्ण भूमिका निभाई, प्रत्येक चरण में अनिवार्य साइन-ऑफ को लागू करते हुए और 21 CFR भाग 11 आवश्यकताओं के अनुसार अपरिवर्तनीय ऑडिट ट्रेल्स को बनाए रखते हुए [3][2].
ये कठोर प्रथाएं स्वाभाविक रूप से इस बात में विस्तारित हुईं कि कंपनियां विचलन और विफलताओं को कैसे संभालती थीं।
बैच विफलताओं के लिए CAPA प्रक्रियाएं
जब बैच विनिर्देशों को पूरा करने में विफल रहे, तो FDA-स्वीकृत कंपनियों ने तेजी से और व्यवस्थित कार्रवाई की। उनकी सुधारात्मक और निवारक कार्रवाई (CAPA) प्रक्रियाओं में औपचारिक मूल कारण विश्लेषण, प्रभाव आकलन, और स्पष्ट रूप से प्रलेखित सुधारात्मक कार्रवाइयाँ शामिल थीं [3]. किसी भी विचलन को एकीकृत गुणवत्ता आश्वासन ढांचे के भीतर प्रबंधित किया गया, यह सुनिश्चित करते हुए कि सभी मुद्दों की पूरी तरह से जांच, औचित्य, और प्रलेखन किया गया था, इससे पहले कि उत्पादन जारी रहा [2].
आगे देखते हुए, डेटा अखंडता 2024–2025 के लिए FDA प्रवर्तन कार्यों का एक प्रमुख फोकस बनने के लिए तैयार है [1].
अपने बैच रिकॉर्ड प्रथाओं को कैसे सुधारें
बैच रिकॉर्ड प्रथाओं को मजबूत करने के लिए सटीक दस्तावेज़ीकरण की आवश्यकता होती है ताकि FDA निरीक्षणों के दौरान अक्सर पहचानी जाने वाली सामान्य विफलताओं को संबोधित किया जा सके। यहां कुछ रणनीतियाँ दी गई हैं जो प्रमुख चुनौतियों का सामना करने में मदद कर सकती हैं।
स्वतंत्र बैच रिकॉर्ड ऑडिट का संचालन करें
नियमित तृतीय-पक्ष ऑडिट उन मुद्दों को उजागर कर सकते हैं जिन्हें आंतरिक समीक्षाएं नजरअंदाज कर सकती हैं। प्रयोगशाला सूचना प्रबंधन प्रणाली (LIMS), विनिर्माण निष्पादन प्रणाली (MES), और उद्यम संसाधन योजना (ERP) जैसे महत्वपूर्ण प्रणालियों पर ध्यान केंद्रित करके शुरू करें। रिलीज़ परीक्षण रिकॉर्ड, स्थिरता डेटा, और बैच उत्पादन रिकॉर्ड जैसे उच्च नियामक प्रभाव वाले दस्तावेज़ों को प्राथमिकता दें।
एक प्रभावी विधि नमूना पुनः प्राप्ति परीक्षण है।हाल के बैचों को यादृच्छिक रूप से चुनें और उनके उत्पादन और प्रयोगशाला इतिहास का पुनर्निर्माण करें। यह लापता डेटा, अधूरी हस्ताक्षर, या दस्तावेज़ीकरण अंतराल को इंगित करने में मदद कर सकता है जो नियामक उद्धरणों का कारण बन सकते हैं। अनधिकृत परिवर्तनों या हटाने की पहचान करने के लिए मैन्युअल प्रविष्टियों के साथ सिस्टम-जनित ऑडिट ट्रेल्स का क्रॉस-चेक करें।
पिछले वर्ष की सभी आउट-ऑफ-स्पेसिफिकेशन (OOS) और आउट-ऑफ-ट्रेंड (OOT) रिपोर्टों की समीक्षा करें। मूल्यांकन करें कि क्या मूल कारण विश्लेषण गहन थे और क्या सुधारात्मक और निवारक क्रियाएं (CAPAs) पर्याप्त रूप से लागू की गई थीं। यह ध्यान देने योग्य है कि दस्तावेज़ीकरण मुद्दे FDA चेतावनी पत्रों का 21% हिस्सा बनाते हैं, जबकि मानव त्रुटि फार्मास्युटिकल निर्माण में बैच रिकॉर्ड समस्याओं का 50% योगदान देती है [2].
"यह प्रक्रिया की जटिलता नहीं है जो उद्धरणों को ट्रिगर करती है - यह असंगति, अपूर्णता, और खराब निगरानी है।" – GXP ऑडिटिंग & परामर्श सेवाएँ [5]
नियमित मॉक समीक्षाओं के माध्यम से नियामक निरीक्षणों का अनुकरण करें। यह अभ्यास टीमों को वास्तविक ऑडिट से पहले असंगतियों और संभावित डेटा अखंडता मुद्दों को पहचानने में मदद करता है। सुनिश्चित करें कि सभी रिकॉर्ड ALCOA+ सिद्धांतों का पालन करते हैं: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, और Available।
एक बार जब दस्तावेज़ीकरण की अखंडता मजबूत हो जाए, तो सभी उत्पादन इनपुट्स की गुणवत्ता की पुष्टि करने पर ध्यान केंद्रित करें।
सभी इनपुट्स का माइक्रोबायोलॉजिकल संदूषण के लिए परीक्षण करें
सभी इनपुट्स के लिए स्वतंत्र नसबंदी और शक्ति परीक्षण आवश्यक है - केवल आपूर्तिकर्ता के विश्लेषण प्रमाणपत्रों (CoAs) पर निर्भर न रहें। यह विशेष रूप से संवर्धित मांस उत्पादकों के लिए महत्वपूर्ण है, क्योंकि संदूषण पूरे बैचों को खतरे में डाल सकता है।
उदाहरण के लिए, फरवरी 2013 में, सेंट्रल एडमिक्सचर फार्मेसी सर्विसेज को बाँझ उत्पादों के बैच रिलीज के दौरान अपर्याप्त सूक्ष्मजीव नियंत्रण के कारण FDA के उद्धरणों का सामना करना पड़ा। कंपनी को अपने मानक संचालन प्रक्रियाओं (SOPs) में विस्तृत सूक्ष्मजीव नियंत्रण प्रक्रियाओं को शामिल करना पड़ा[4].
प्रक्रिया में सूक्ष्मजीव चेकपॉइंट्स अंतिम उत्पाद परीक्षण पर अत्यधिक निर्भरता को रोक सकते हैं। इन चेकपॉइंट्स को बैच रिलीज SOPs में शामिल करें और सख्त समकालीन दस्तावेज़ीकरण बनाए रखें। सभी परीक्षण परिणामों और निर्माण चरणों को उसी समय रिकॉर्ड करें ताकि बैक-डेटिंग या विलंबित प्रविष्टियों से बचा जा सके, जो FDA उद्धरणों का कारण बन सकते हैं।
विस्तृत आपूर्तिकर्ता फाइलें रखें, जिनमें CoAs, ऑडिट रिपोर्ट, गुणवत्ता समझौते, और आने वाली सामग्रियों से संबंधित किसी भी विचलन का इतिहास शामिल हो।
बैच रिकॉर्ड प्रथाओं को मजबूत करना आगे की प्रक्रिया को HACCP और GCCP जैसे स्थापित मानकों के साथ संरेखित करना शामिल करता है।
HACCP और GCCP मानकों के साथ रिकॉर्ड्स को संरेखित करें
बैच रिकॉर्ड्स में Hazard Analysis Critical Control Point (HACCP) सिद्धांतों को शामिल करने से यह सुनिश्चित होता है कि उत्पादन के दौरान महत्वपूर्ण प्रक्रिया चर की निगरानी और दस्तावेजीकरण किया जाता है। इसमें अंतिम चरण के परीक्षणों पर निर्भर रहने के बजाय इन-प्रोसेस माइक्रोबियल परीक्षण चेकपॉइंट्स की स्थापना शामिल है।
संवर्धित मांस उत्पादकों के लिए, Good Cell Culture Practice (GCCP) मानकों का पालन करना समान रूप से महत्वपूर्ण है। बैच रिकॉर्ड्स में एसेप्टिक हेरफेर, गाउनिंग प्रक्रियाओं और बैच रिलीज़ मानदंडों से जुड़े पर्यावरणीय निगरानी का विवरण शामिल होना चाहिए[3][4]. ये कदम अनुपालन बनाए रखने और उत्पाद की सुरक्षा सुनिश्चित करने में मदद करते हैं।
उद्योग डेटा दिखाता है कि 52% दस्तावेज़ीकरण उल्लंघन तब बढ़ जाते हैं जब उचित बैच निर्माण सॉफ़्टवेयर मौजूद नहीं होता[3][4]. एक उदाहरण: फरवरी 2023 में, नेफ्रॉन स्टेराइल कंपाउंडिंग सेंटर को बैच रिलीज़ से पहले महत्वपूर्ण प्रक्रिया वेरिएबल्स को सत्यापित करने के लिए नियंत्रण प्रक्रियाओं की अनुपस्थिति के कारण एफडीए अवलोकन प्राप्त हुआ [4]. यह मान्यता प्राप्त मानकों के साथ संरेखित सक्रिय दस्तावेज़ीकरण की आवश्यकता को उजागर करता है।
इलेक्ट्रॉनिक बैच रिकॉर्ड्स (EBR) में संक्रमण दस्तावेज़ीकरण त्रुटियों को काफी हद तक कम कर सकता है - वास्तविक समय डेटा संग्रह और स्वचालित वर्कफ़्लोज़ के माध्यम से 50% तक - [2]. ये सिस्टम एक बैच के आगे बढ़ने से पहले गायब माइक्रोबियल परीक्षण परिणामों या अधूरी समीक्षाओं को चिह्नित करते हैं, जिससे मानव त्रुटि कम होती है।
"एफडीए अपेक्षा करता है कि रिकॉर्ड्स ALCOA(+) हों: Attributable, Legible, Contemporaneous, Original, Accurate - साथ ही Complete, Consistent, Enduring, और Available।" – एटलस अनुपालन [1]
बैच रिकॉर्ड्स में हर अस्पष्ट विसंगति या विचलन को औपचारिक जांच और CAPA प्रणाली से जोड़ा जाना चाहिए। इलेक्ट्रॉनिक माइक्रोबायोलॉजिकल परीक्षण डेटा की अखंडता की सुरक्षा के लिए लिखने और हटाने की अनुमतियों को सीमित करें। प्रतिस्पर्धी निर्माता उत्पादन पूर्णता के 30 दिनों के भीतर 95% बैचों की समीक्षा और रिलीज़ करने का लक्ष्य रखते हैं [2].
ये क्रियाएं न केवल उद्धरणों के जोखिम को कम करती हैं बल्कि हाल की FDA निरीक्षणों में उजागर किए गए कठोर दस्तावेज़ीकरण मानकों के साथ भी मेल खाती हैं।
बायोफार्मा बनाम संवर्धित मांस: बैच रिकॉर्ड अंतर
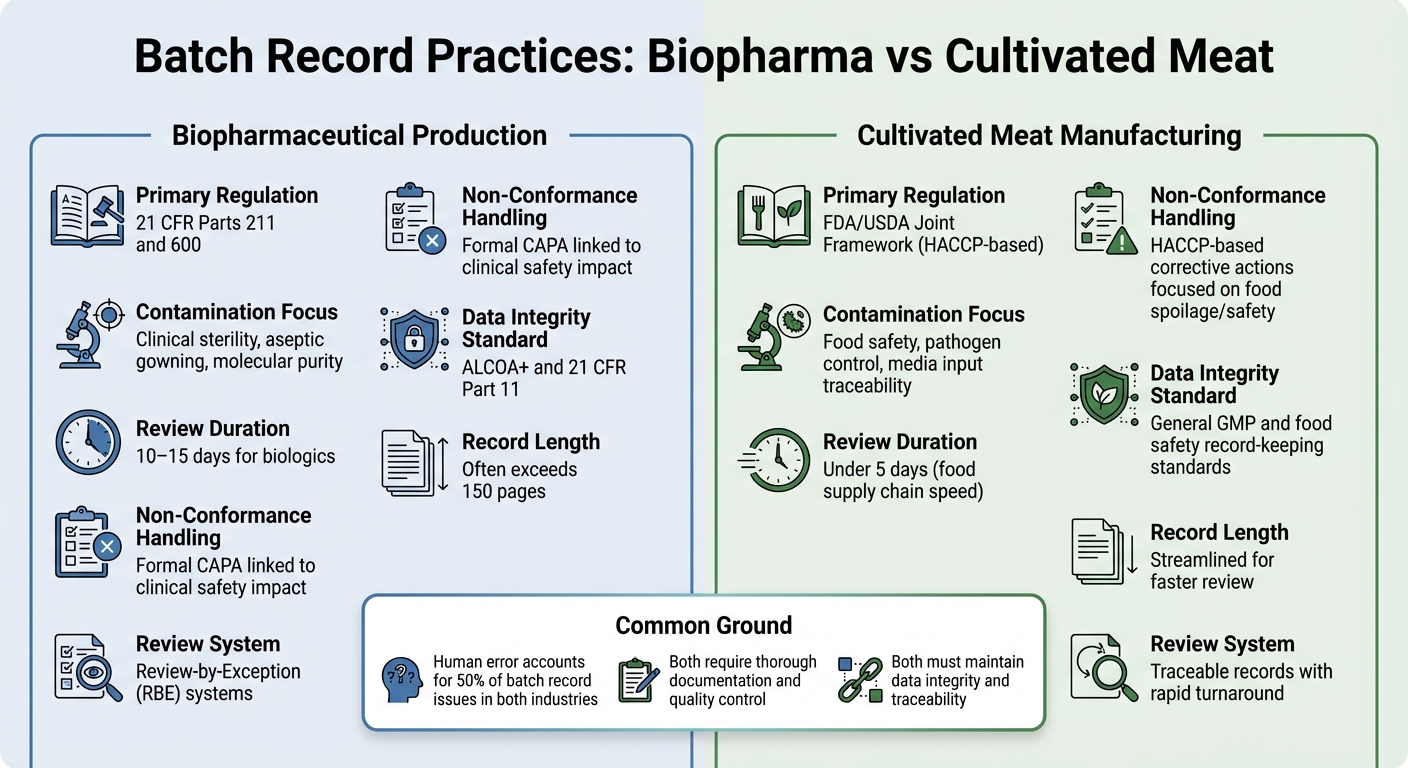
बायोफार्मास्यूटिकल बनाम संवर्धित मांस बैच रिकॉर्ड आवश्यकताओं की तुलना
बायोफार्मास्यूटिकल उत्पादन और संवर्धित मांस निर्माण के बीच बैच रिकॉर्ड प्रथाओं में अंतर को देखना इन उद्योगों में नियामक मांगों के दस्तावेजीकरण प्राथमिकताओं को कैसे आकार देता है, इसका एक स्पष्ट चित्र प्रदान करता है।
दोनों क्षेत्रों को विस्तृत दस्तावेजीकरण की आवश्यकता होती है, लेकिन उनके नियामक ढांचे और नियंत्रण उद्देश्यों में काफी अंतर होता है। बायोफार्मा में, बैच रिकॉर्ड 21 CFR भाग 211 और 600, के तहत सख्ती से विनियमित होते हैं, जो गुणवत्ता नियंत्रण इकाई को बैच जारी करने से पहले सभी उत्पादन और नियंत्रण रिकॉर्ड की समीक्षा और अनुमोदन करने की आवश्यकता होती है [2]. संवर्धित मांस उत्पादक, दूसरी ओर, आमतौर पर HACCP और GCCP मानकों का पालन करते हैं।ये खाद्य सुरक्षा और रोगजनक नियंत्रण पर अधिक ध्यान केंद्रित करते हैं, बजाय इसके कि इंजेक्टेबल बायोलॉजिक्स के लिए मांगी गई क्लिनिकल-ग्रेड नसबंदी पर।
बायोफार्मा बैच रिकॉर्ड अक्सर विस्तृत होते हैं, कभी-कभी 150 पृष्ठों से अधिक होते हैं, और समीक्षा प्रक्रिया में 10-15 दिन लग सकते हैं। इसे सरल बनाने के लिए, कई बायोफार्मा कंपनियां Review-by-Exception (RBE) सिस्टम का उपयोग करती हैं, जो प्रमुख विचलनों को एक संक्षिप्त रिपोर्ट में संक्षेपित करती हैं। इस बीच, संवर्धित मांस उत्पादक ऐसे रिकॉर्ड का लक्ष्य रखते हैं जिन्हें पांच दिनों से कम समय में समीक्षा किया जा सके, जो खाद्य आपूर्ति श्रृंखला की तेज गति को दर्शाता है [2].
इन रिकॉर्ड की सामग्री भी विभिन्न प्राथमिकताओं को उजागर करती है। बायोफार्मा निरीक्षण अक्सर एसेप्टिक प्रोसेसिंग विवरणों पर ध्यान केंद्रित करते हैं, जैसे कि गाउनिंग प्रक्रियाएं और पर्यावरणीय नियंत्रण। इसके विपरीत, संवर्धित मांस रिकॉर्ड को खाद्य सुरक्षा सुनिश्चित करने के लिए मीडिया इनपुट और सूक्ष्मजीवविज्ञान परीक्षण पर जोर देना चाहिए।संवर्धित मांस के लिए, चुनौती जटिल मीडिया इनपुट्स को ट्रैक करने, सभी सामग्रियों के लिए सूक्ष्मजीवविज्ञान परीक्षणों का दस्तावेजीकरण करने, और खाद्य सुरक्षा के महत्वपूर्ण सीमाओं को पूरा करने में है - बिना फार्मास्यूटिकल्स की सख्त नसबंदी आवश्यकताओं का पालन किए।
संक्रमण और गैर-अनुरूपता प्रवृत्तियाँ
| विशेषता | बायोफार्मास्यूटिकल उत्पादन | संवर्धित मांस निर्माण |
|---|---|---|
| प्राथमिक विनियमन | 21 CFR भाग 211 और 600[2] | FDA/USDA संयुक्त ढांचा (HACCP-आधारित) |
| संक्रमण फोकस | क्लिनिकल नसबंदी, एसेप्टिक गाउनिंग, आणविक शुद्धता[2] | खाद्य सुरक्षा, रोगजनक नियंत्रण, मीडिया इनपुट ट्रेसबिलिटी |
| समीक्षा अवधि | बायोलॉजिक्स के लिए 10–15 दिन[2] | 5 दिनों के भीतर (खाद्य आपूर्ति श्रृंखला की गति) |
| गैर-अनुरूपता प्रबंधन | क्लिनिकल सुरक्षा प्रभाव से जुड़ा औपचारिक CAPA [2] | खाद्य खराबी/सुरक्षा पर केंद्रित HACCP-आधारित सुधारात्मक क्रियाएँ |
| डेटा अखंडता मानक | ALCOA+ और 21 CFR भाग 11 [1] | सामान्य GMP और खाद्य सुरक्षा रिकॉर्ड-रखरखाव मानक |
जबकि मानव त्रुटि दर दोनों उद्योगों में समान है - लगभग 50% बैच रिकॉर्ड मुद्दे मानव गलतियों से उत्पन्न होते हैं [2] - दांव अलग हैं।बायोफार्मा में, एक भी अप्रलेखित विचलन रोगी सुरक्षा के लिए गंभीर प्रभाव डाल सकता है। संवर्धित मांस के लिए, संदूषण के जोखिम खाद्यजनित रोगाणुओं और खराबी के बारे में अधिक होते हैं, जो पूरे उत्पादन रन को प्रभावित कर सकते हैं।
निष्कर्ष
बैच रिकॉर्ड हर संवर्धित मांस उत्पादन रन के लिए आधिकारिक लॉग के रूप में कार्य करते हैं - यदि कोई कदम दर्ज नहीं किया गया है, तो नियामक इसे निष्पादित नहीं मानते [6][3]. यह सटीक दस्तावेजीकरण और सख्त गुणवत्ता नियंत्रण के महत्व को उजागर करता है।
एफडीए निरीक्षण इस बात पर जोर देते हैं कि डेटा अखंडता को ALCOA+ सिद्धांतों के साथ संरेखित होना चाहिए [1]. गुणवत्ता नियंत्रण टीमों को आवश्यक है कि वे सभी उत्पादन रिकॉर्ड की समीक्षा और अनुमोदन करें, इससे पहले कि एक बैच जारी किया जा सके [2][17], और किसी भी विचलन की तुरंत जांच की जानी चाहिए और एक दस्तावेजीकृत मूल कारण विश्लेषण के साथ [2][5]. जबकि मानव त्रुटि बैच रिकॉर्ड मुद्दों के 50% के लिए जिम्मेदार है, दो-स्तरीय समीक्षाएं और संरचित CAPA (सुधारात्मक और निवारक कार्रवाई) प्रक्रियाएं इन जोखिमों को कम करने में मदद कर सकती हैं [2][5].
"यह प्रक्रिया की जटिलता नहीं है जो उद्धरणों को ट्रिगर करती है - यह असंगति, अपूर्णता, और खराब निगरानी है।" - GXP ऑडिटिंग & परामर्श सेवाएँ [5]
इन चुनौतियों को पार करने के लिए, संवर्धित मांस उत्पादकों को स्वतंत्र ऑडिट, खाद्य-सुरक्षित सामग्री के माइक्रोबायोलॉजिकल संदूषण के लिए कठोर परीक्षण, और यह सुनिश्चित करने पर ध्यान केंद्रित करना चाहिए कि दस्तावेज़ीकरण HACCP और GCCP मानकों का पालन करता है। इलेक्ट्रॉनिक बैच रिकॉर्ड सिस्टम को लागू करना, जो 21 CFR भाग 11 [1], के तहत सत्यापित है, त्रुटियों को काफी हद तक कम कर सकता है और समीक्षा प्रक्रियाओं को तेज कर सकता है।
नियामक वातावरण सटीकता की मांग करता है, लेकिन यह नेविगेट करने योग्य है।बायोफार्मा की गलतियों से सीखकर - जैसे कि Qinhuangdao Zizhu Pharmaceutical [17], पर हस्ताक्षर की कमी, Terumo Corp [18] , पर अपर्याप्त द्वैध सत्यापन और Torrent Pharmaceuticals [18] पर अपर्याप्त विचलन दस्तावेजीकरण - संवर्धित मांस कंपनियाँ शुरुआत से ही अनुपालन प्रणाली स्थापित कर सकती हैं। इन पाठों को शामिल करने से सक्रिय अनुपालन और निरंतर गुणवत्ता सुनिश्चित होती है। सुरक्षित रिकॉर्ड संरक्षण, समय पर विचलन रिपोर्टिंग, और यथार्थवादी मॉक ऑडिट का संचालन यह सुनिश्चित करेगा कि बैच रिकॉर्ड निरीक्षण के लिए तैयार रहें और उत्पादन रन पूरी तरह से ट्रेस करने योग्य हों।
संवर्धित मांस निर्माण में उच्च उत्पादन मानकों को बनाए रखने के लिए अधिक संसाधनों और विशेषज्ञ मार्गदर्शन के लिए,
सामान्य प्रश्न
संवर्धित मांस के लिए एक बैच रिकॉर्ड में क्या शामिल होना चाहिए?
संवर्धित मांस के लिए एक बैच रिकॉर्ड पूरे निर्माण प्रक्रिया का एक व्यापक लॉग होता है। इसमें शामिल होना चाहिए विस्तृत प्रसंस्करण निर्देश, चरण-दर-चरण निष्पादन रिकॉर्ड, और उत्पादन के दौरान होने वाले किसी भी विचलन को नोट करें। इसके अतिरिक्त, इसे प्रक्रिया में परीक्षण और रिलीज परीक्षण का दस्तावेजीकरण करना चाहिए ताकि यह सुनिश्चित हो सके कि उत्पाद सुरक्षा, गुणवत्ता और नियामक मानकों को पूरा करता है।
हम बैच रिकॉर्ड का उपयोग करके नसबंदी कैसे साबित कर सकते हैं?
बैच रिकॉर्ड के माध्यम से नसबंदी साबित करना दस्तावेजीकृत नसबंदी प्रक्रियाओं, परीक्षण परिणामों और मीडिया गुणवत्ता नियंत्रण रिपोर्टों की पूरी तरह से जांच करना शामिल है ताकि यह सुनिश्चित किया जा सके कि वे नियामक आवश्यकताओं को पूरा करते हैं।यह महत्वपूर्ण है कि किसी भी विचलन या असफल परीक्षणों को विस्तृत जांच और CAPAs (सुधारात्मक और निवारक क्रियाएं) के माध्यम से संबोधित किया जाए। यह प्रक्रिया सुनिश्चित करती है कि हर कदम का पालन किया गया है और किसी भी समस्या को ठीक से हल किया गया है ताकि स्वच्छता मानकों को बनाए रखा जा सके।
इलेक्ट्रॉनिक बैच रिकॉर्ड्स (भाग 11) की आवश्यकता कब होती है?
इलेक्ट्रॉनिक बैच रिकॉर्ड्स भाग 11 के तहत आवश्यक होते हैं जब इलेक्ट्रॉनिक सिस्टम का उपयोग बैच रिकॉर्ड विचलनों को दस्तावेज़, जांच और न्यायसंगत बनाने के लिए किया जाता है। वे 21 CFR Part 211.192 , के अनुपालन को सुनिश्चित करने में महत्वपूर्ण भूमिका निभाते हैं, डेटा अखंडता की सुरक्षा करते हैं, जांच समयसीमा को पूरा करते हैं, और प्रभावी प्रबंधन निगरानी सुनिश्चित करते हैं।









