

ในห้องปลอดเชื้อ การปฏิบัติตามมาตรฐาน GMP ช่วยให้มั่นใจในคุณภาพและความปลอดภัยที่สม่ำเสมอโดยการกำหนดให้มีการตรวจสอบอย่างละเอียดและบันทึกข้อมูลที่ถูกต้อง สำหรับโรงงานผลิตเนื้อสัตว์เพาะเลี้ยง สิ่งนี้มีความสำคัญอย่างยิ่ง เนื่องจากแม้แต่การเบี่ยงเบนเล็กน้อยในสภาพห้องปลอดเชื้อก็อาจส่งผลต่อการเจริญเติบโตของเซลล์หรือทำให้การผลิตปนเปื้อนได้
ประเด็นสำคัญ:
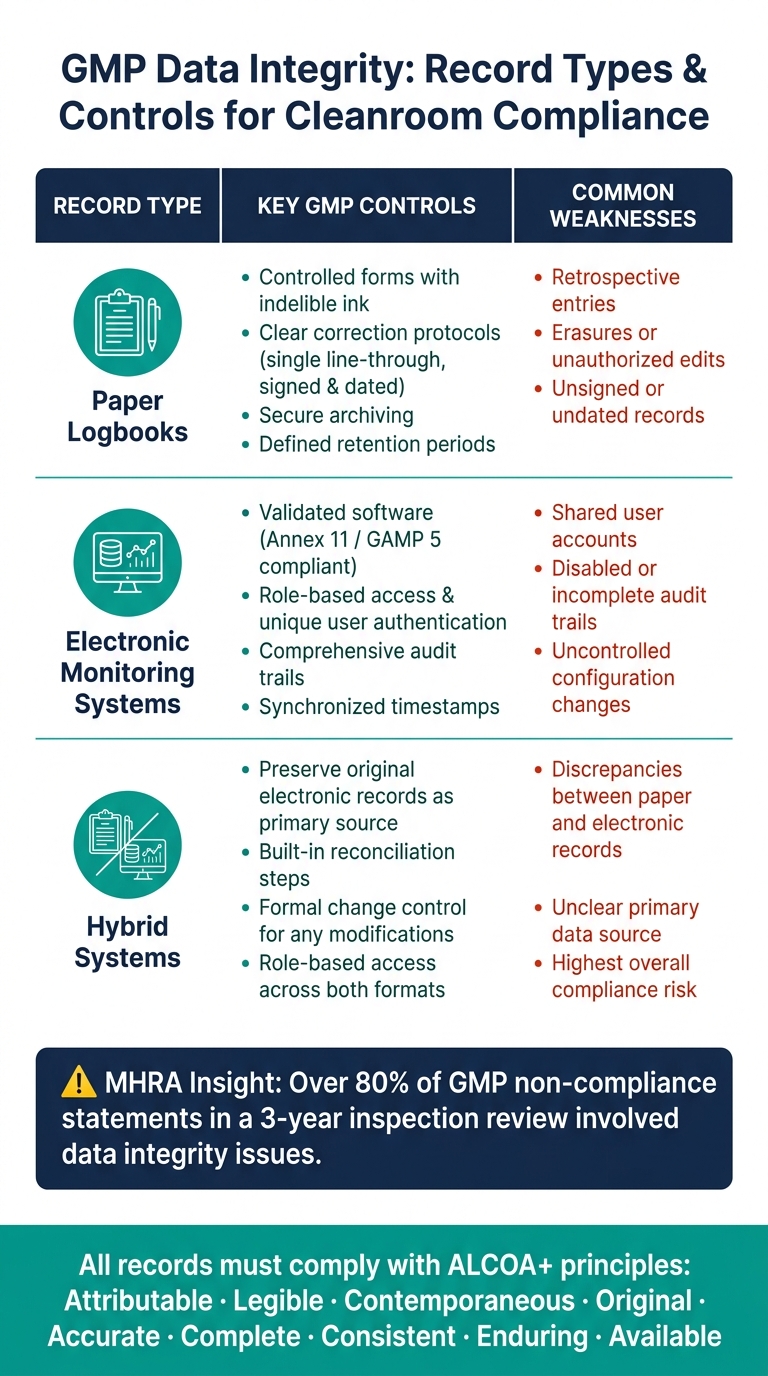
- มาตรฐาน GMP: เน้นความสมบูรณ์ของข้อมูล โดยปฏิบัติตามกรอบงาน ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available).
- พารามิเตอร์ที่สำคัญ: ตรวจสอบอนุภาคในอากาศ จำนวนจุลินทรีย์ อุณหภูมิ ความชื้น และความดันเพื่อตรวจจับความเสี่ยงตั้งแต่เนิ่นๆ ซึ่งต้องการ การเลือกเซ็นเซอร์ที่แม่นยำ ที่สามารถรักษาพารามิเตอร์ที่สำคัญเหล่านี้ได้
- ระบบข้อมูล: ใช้ ระบบควบคุมกระบวนการชีวภาพที่ผ่านการตรวจสอบแล้ว พร้อมการเข้าถึงตามบทบาท เส้นทางการตรวจสอบ และการจัดเก็บที่ปลอดภัยสำหรับทั้งบันทึกอิเล็กทรอนิกส์และกระดาษ
- ความเสี่ยงทั่วไป: หลีกเลี่ยงข้อผิดพลาดในการจัดการข้อมูลด้วยตนเอง การเปลี่ยนแปลงการกำหนดค่าโดยไม่มีการควบคุม และการปฏิบัติในการจัดเก็บที่ไม่เหมาะสม
- GMP ที่ปรับให้เหมาะสมสำหรับเนื้อสัตว์ที่เพาะเลี้ยง: ปรับกลยุทธ์การตรวจสอบเพื่อตอบสนองความเสี่ยงเฉพาะ เช่น สภาพของเครื่องปฏิกรณ์ชีวภาพและสารตกค้างจากสารทำความสะอาด
สำหรับการวิจัยและพัฒนาเนื้อสัตว์ที่เพาะเลี้ยง การจัดการข้อมูลที่แข็งแกร่งช่วยให้มั่นใจในความปลอดภัยของผลิตภัณฑ์ การปฏิบัติตามกฎระเบียบ และการดำเนินงานที่สามารถขยายได้ จัดการกับช่องโหว่ที่ทราบล่วงหน้าเพื่อหลีกเลี่ยงปัญหาด้านกฎระเบียบที่มีค่าใช้จ่ายสูงในภายหลัง
ข้อกำหนด GMP ที่สำคัญสำหรับความสมบูรณ์ของข้อมูลในห้องสะอาด
การทำความเข้าใจหลักการ ALCOA+
รากฐานของความสมบูรณ์ของข้อมูล GMP อยู่ในกรอบงาน ALCOA+ หน่วยงานกำกับดูแล เช่น MHRA , EMA, และ WHO ใช้เพื่อพิจารณาว่าบันทึกในห้องสะอาดสามารถเชื่อถือได้หรือไม่ALCOA+ ย่อมาจาก: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, และ Available . แต่ละคำเหล่านี้มีความสำคัญในทางปฏิบัติในการดำเนินงานในห้องปลอดเชื้อ.
- Attributable: ทุกการบันทึก - ไม่ว่าจะเป็นการนับอนุภาค, การอ่านค่าความดัน, หรือบันทึกการทำความสะอาด - ต้องแสดงให้ชัดเจนว่าใครบันทึก พร้อมวันที่, เวลา, และรายละเอียดเครื่องมือที่เกี่ยวข้อง.
- Legible: บันทึกต้องอ่านง่ายและเข้าใจได้ เพื่อความชัดเจนในระหว่างการตรวจสอบหรือการตรวจเยี่ยม.
- Contemporaneous: ข้อมูลต้องถูกบันทึกในเวลาจริง การบันทึกที่ล่าช้าหรือย้อนหลังอาจทำให้ความน่าเชื่อถือของบันทึกเสียหาย.
- Original: ข้อมูลควรคงอยู่ในรูปแบบที่บันทึกครั้งแรก โดยไม่มีการแก้ไขหรือเปลี่ยนแปลงโดยไม่ได้รับอนุญาต.
- ถูกต้อง: ค่าที่บันทึกต้องสะท้อนผลลัพธ์ที่สังเกตได้อย่างแท้จริง ปราศจากข้อผิดพลาดหรือการบิดเบือน
- ครบถ้วน: รายการที่เกี่ยวข้องทั้งหมด รวมถึงการเบี่ยงเบนหรือผลลัพธ์ที่อยู่นอกข้อกำหนด ต้องได้รับการบันทึก
- สม่ำเสมอ, ยั่งยืน, และ พร้อมใช้งาน: บันทึกควรเป็นไปตามลำดับที่ถูกต้อง ถูกเก็บรักษาไว้อย่างครบถ้วนตามระยะเวลาที่กำหนด และสามารถเข้าถึงได้ง่ายสำหรับการตรวจสอบหรือการตรวจสอบ
หน่วยงานกำกับดูแลให้ความสำคัญอย่างมากกับหลักการเหล่านี้ ตัวอย่างเช่น การตรวจสอบของ MHRA พบว่ากว่า 80% ของคำแถลงการไม่ปฏิบัติตาม GMP ในช่วงสามปีเกี่ยวข้องกับปัญหาความสมบูรณ์ของข้อมูล [5]. เพื่อฝัง ALCOA+ ในการทำงานประจำวัน สถานที่สามารถใช้แบบฟอร์มที่มีโครงสร้างดี บังคับใช้ฟิลด์ที่จำเป็น และดำเนินการตรวจสอบเส้นทางการตรวจสอบเป็นประจำ
ด้วย ALCOA+ เป็นพื้นฐาน ขั้นตอนต่อไปคือการรับรองว่าหลักการเหล่านี้ถูกยึดถือในระบบกระดาษ อิเล็กทรอนิกส์ และระบบผสม
การรับรองความสมบูรณ์ของข้อมูลในทุกฟอร์แมต
ในโรงงานผลิตเนื้อสัตว์ที่เพาะเลี้ยง ซึ่งข้อมูลมีผลโดยตรงต่อการตัดสินใจปล่อยชุด การรักษาความสมบูรณ์ในทุกรูปแบบของบันทึกเป็นสิ่งที่ไม่สามารถต่อรองได้ GMP กำหนดให้มีความสมบูรณ์ในระดับเดียวกันทั้งบันทึกกระดาษและอิเล็กทรอนิกส์ แม้ว่าการควบคุมเฉพาะอาจแตกต่างกันไปตามรูปแบบ
- ระบบกระดาษ: แนวปฏิบัติที่ดีที่สุดรวมถึงการใช้แบบฟอร์มที่ควบคุมด้วยหมึกที่ไม่สามารถลบได้และโปรโตคอลการแก้ไขที่ชัดเจน (e.g. การแก้ไขด้วยการขีดเส้นเดียวพร้อมลายเซ็นและวันที่) การเก็บรักษาอย่างปลอดภัยและการปฏิบัติตามระยะเวลาการเก็บรักษาที่กำหนดก็มีความสำคัญเช่นกัน
- ระบบอิเล็กทรอนิกส์: ควรทำงานบนซอฟต์แวร์ที่ผ่านการตรวจสอบแล้วซึ่งสอดคล้องกับ Annex 11 และ GAMP 5. คุณสมบัติหลักประกอบด้วยการเข้าถึงตามบทบาท, การยืนยันตัวตนของผู้ใช้ที่ไม่ซ้ำกัน, เส้นทางการตรวจสอบที่ครอบคลุม, และการประทับเวลาที่ซิงโครไนซ์ การตรวจสอบเส้นทางการตรวจสอบเป็นประจำเป็นสิ่งสำคัญในการระบุและแก้ไขความผิดปกติใด ๆ
- ระบบไฮบริด: เหล่านี้เป็นความเสี่ยงสูงสุดเนื่องจากเกี่ยวข้องกับทั้งบันทึกอิเล็กทรอนิกส์และกระดาษ ตัวอย่างเช่น เมื่อเครื่องมือสร้างข้อมูลอิเล็กทรอนิกส์ที่ถูกถ่ายโอนไปยังบันทึกกระดาษ ข้อมูลอิเล็กทรอนิกส์ต้นฉบับต้องถูกเก็บรักษาไว้เป็นบันทึกหลัก ควรมีขั้นตอนการกระทบยอดในกระบวนการทำงานเพื่อตรวจจับและแก้ไขความไม่สอดคล้องกันระหว่างบันทึกอิเล็กทรอนิกส์และกระดาษ ซึ่งมีความสำคัญอย่างยิ่งในการผลิตเนื้อสัตว์ที่เพาะเลี้ยง ซึ่งแม้แต่ความไม่สอดคล้องกันของข้อมูลเล็กน้อยก็อาจทำให้มาตรการควบคุมการปนเปื้อนเสียหายได้มาตรการควบคุมการปนเปื้อน.
ตารางด้านล่างสรุปการควบคุมที่สำคัญและจุดอ่อนทั่วไปสำหรับแต่ละประเภทของบันทึก:
| ประเภทบันทึก | การควบคุม GMP ที่สำคัญ | จุดอ่อนทั่วไป |
|---|---|---|
| แบบฟอร์มที่ควบคุม, หมึกที่ไม่สามารถลบได้, โปรโตคอลการแก้ไขที่ชัดเจน, การลงชื่อและวันที่ในรายการ | การบันทึกย้อนหลัง, การลบ, บันทึกที่ไม่มีลายเซ็น | |
| ระบบการตรวจสอบอิเล็กทรอนิกส์ | ซอฟต์แวร์ที่ผ่านการตรวจสอบ, การเข้าถึงตามบทบาท, เส้นทางการตรวจสอบ, การซิงโครไนซ์เวลา | บัญชีผู้ใช้ที่ใช้ร่วมกัน, เส้นทางการตรวจสอบที่ถูกปิดหรือไม่สมบูรณ์ |
| ระบบไฮบริด | รักษาบันทึกอิเล็กทรอนิกส์ต้นฉบับ; ดำเนินการขั้นตอนการกระทบยอด | ความไม่สอดคล้องกันระหว่างบันทึกกระดาษและอิเล็กทรอนิกส์, แหล่งข้อมูลหลักที่ไม่ชัดเจน |
เพื่อให้เป็นไปตามข้อกำหนด ควรจัดหมวดหมู่บันทึกตามความสำคัญของพวกเขาสำหรับโรงงานผลิตเนื้อสัตว์ที่เพาะเลี้ยง ข้อมูลที่เกี่ยวข้องกับการตัดสินใจปล่อยชุดหรือการควบคุมการปนเปื้อน (e.g. , ผลการตรวจสอบสิ่งแวดล้อม, บันทึกการเตือนภัย HVAC, หรือข้อมูลการทดสอบความสมบูรณ์ของตัวกรอง) ควรอยู่ภายใต้การควบคุมการเข้าถึงที่เข้มงวดที่สุด การตรวจสอบบ่อยครั้ง และการจัดการเส้นทางการตรวจสอบที่แข็งแกร่ง
sbb-itb-ffee270
การตรวจสอบสิ่งแวดล้อมประจำในห้องสะอาด GMP & ความสมบูรณ์ของข้อมูล 21CFR part 11
การจัดการวงจรชีวิตข้อมูลในห้องสะอาด
ความสมบูรณ์ของข้อมูล GMP: ประเภทของบันทึก & การควบคุมเพื่อความสอดคล้องในห้องสะอาด
ขั้นตอนของวงจรชีวิตข้อมูลในห้องสะอาด
วงจรชีวิตข้อมูลในห้องสะอาดในโรงงานผลิตเนื้อสัตว์ที่เพาะเลี้ยงประกอบด้วยหลายขั้นตอน แต่ละขั้นตอนมีข้อกำหนดการปฏิบัติตามที่เฉพาะเจาะจง
การสร้างข้อมูล เป็นจุดเริ่มต้น This includes readings from instruments like particle counters, differential pressure sensors, temperature and humidity probes, viable air samplers, surface contact plates, and cleaning verification logs. For each parameter, there must be a documented sampling frequency, a designated operator, and a calibrated instrument. Aligning these monitoring tasks with production stages - such as inoculation, cell expansion, or harvest - helps demonstrate how environmental control links directly to product quality and safety.
Once generated, data enters the capture and transfer stage. Ideally, electronic systems should automatically record readings with time-stamped entries tied to individual user accounts. For paper-based entries, data must be logged in real time using indelible ink, with reconciliation checks in place when transferring the data to electronic systems.
The storage phase is equally critical. ทั้งข้อมูลดิบและข้อมูลที่ผ่านการประมวลผลแล้วต้องได้รับการเก็บรักษาเพื่อให้สามารถติดตามค่าที่รายงานกลับไปยังบันทึกต้นฉบับได้ ซึ่งต้องการที่เก็บข้อมูลที่ปลอดภัยและผ่านการตรวจสอบ พร้อมการควบคุมการเข้าถึงตามบทบาทและการทดสอบการสำรองข้อมูลเป็นประจำ การสำรองข้อมูลควรเก็บไว้ในสถานที่ที่แยกจากระบบหลักและตรวจสอบเป็นระยะเพื่อให้แน่ใจว่าสามารถกู้คืนได้เมื่อจำเป็น
สุดท้าย การเก็บถาวร สรุปวงจรชีวิต บันทึกจะเปลี่ยนไปสู่สถานะการเข้าถึงที่ควบคุมได้และอ่านได้อย่างเดียวเมื่อไม่ได้ใช้งานอย่างต่อเนื่อง แต่ต้องสามารถเรียกคืนได้ในช่วงเวลาที่ต้องการเก็บรักษา ในโรงงานผลิตเนื้อสัตว์ที่เพาะเลี้ยง การเก็บถาวรข้อมูลในระยะพัฒนาสามารถสนับสนุนความพยายามในการตรวจสอบในอนาคตได้เช่นกัน
ความเข้าใจที่ชัดเจนเกี่ยวกับขั้นตอนเหล่านี้เป็นสิ่งสำคัญในการจัดการความเสี่ยงอย่างมีประสิทธิภาพ ดังที่ระบุไว้ด้านล่าง
ความเสี่ยงทั่วไปในการจัดการข้อมูล
การจัดการข้อมูลระหว่างการถ่ายโอนมีความเสี่ยงอย่างมากข้อผิดพลาดในการถอดความด้วยตนเองและการป้อนข้อมูลย้อนหลังสามารถบั่นทอนความสมบูรณ์ของข้อมูลได้ เพื่อหลีกเลี่ยงสิ่งนี้ ข้อมูลทั้งหมดต้องปฏิบัติตามหลักการ ALCOA+ (สามารถระบุได้, อ่านออก, ทันเวลา, ต้นฉบับ, ถูกต้อง, รวมถึงความสมบูรณ์, ความสม่ำเสมอ, ความยั่งยืน, และความพร้อมใช้งาน) ในเวลาจริง
การเปลี่ยนแปลงการกำหนดค่า เป็นอีกหนึ่งข้อกังวลหลัก การปรับเปลี่ยนขีดจำกัดการเตือน, การจับคู่เซ็นเซอร์, หรือการตั้งค่านาฬิกาของระบบโดยไม่มีการควบคุมการเปลี่ยนแปลงอย่างเป็นทางการสามารถทำลายความน่าเชื่อถือของข้อมูลที่บันทึกไว้ก่อนและหลังการเปลี่ยนแปลง นอกจากนี้ ความล้มเหลวในการจัดเก็บข้อมูล - ไม่ว่าจะเกิดจากฐานข้อมูลที่เสียหาย, การสำรองข้อมูลที่ไม่ได้ทดสอบ, หรือเอกสารที่เสียหายจากปัจจัยสิ่งแวดล้อม - สามารถทำให้บันทึกที่สำคัญไม่สามารถเข้าถึงได้ เพื่อบรรเทาความเสี่ยงเหล่านี้ ให้แน่ใจว่าทุกกระแสข้อมูลถูกจับคู่กับจุดเก็บถาวรที่กำหนดไว้พร้อมกับการระบุความเป็นเจ้าของอย่างชัดเจน เพื่อลดโอกาสที่ช่องโหว่จะถูกตั้งข้อสังเกตในระหว่างการตรวจสอบตามกฎระเบียบ
การควบคุม GMP สำหรับระบบการตรวจสอบห้องสะอาด
การควบคุมที่สำคัญสำหรับระบบการตรวจสอบ
ระบบการตรวจสอบที่เป็นไปตามมาตรฐาน GMP ต้องพึ่งพาเซ็นเซอร์ที่ผ่านการสอบเทียบ การจัดการข้อมูลที่ปลอดภัย และการจัดการสัญญาณเตือนที่มีประสิทธิภาพ เซ็นเซอร์สำหรับพารามิเตอร์เช่น อุณหภูมิ ความชื้นสัมพัทธ์ ความดันต่างๆ การนับอนุภาคที่ไม่สามารถมีชีวิต และการสุ่มตัวอย่างจุลชีพที่มีชีวิตต้องได้รับการสอบเทียบตามตารางเวลาที่บันทึกไว้และสามารถตรวจสอบย้อนกลับไปยังมาตรฐานที่ได้รับการยอมรับ การโอนข้อมูลอัตโนมัติจากเซ็นเซอร์เหล่านี้พร้อมกับการประทับเวลาที่ซิงโครไนซ์ช่วยลดความเสี่ยงของข้อผิดพลาดจากการทำงานด้วยมือ
การจัดการสัญญาณเตือนมีความสำคัญเท่าเทียมกัน ขีดจำกัดของสัญญาณเตือนควรสอดคล้องกับกรอบการกำกับดูแล เช่น ISO 14644-1 ขีดจำกัดระดับชั้นและคำแนะนำ EU GMP Annex 1 ทุกสัญญาณเตือนที่ถูกกระตุ้นต้องมีการบันทึกการตอบสนอง รวมถึงรายละเอียดของผู้ใช้ การประทับเวลา และความคิดเห็นใดๆการไม่บันทึกการตอบสนองต่อสัญญาณเตือนสร้างช่องโหว่ในการปฏิบัติตามข้อกำหนด
การควบคุมการเข้าถึงตามบทบาทต้องบังคับใช้อย่างเคร่งครัดทั่วทั้งระบบ การอนุญาตในระดับผู้ดูแลระบบควรเป็นสิ่งจำเป็นสำหรับการเปลี่ยนแปลงขีดจำกัดการเตือน การกำหนดค่าตัวเซ็นเซอร์ หรือการตั้งค่านาฬิกาของระบบ และการเปลี่ยนแปลงเหล่านี้ต้องเป็นไปตามกระบวนการควบคุมการเปลี่ยนแปลงอย่างเป็นทางการ จำเป็นต้องมีเส้นทางการตรวจสอบสำหรับการดำเนินการที่เกี่ยวข้องกับ GMP ทั้งหมด เช่น การอัปเดตการกำหนดค่า การลบข้อมูล ลายเซ็นอิเล็กทรอนิกส์ และการปรับเซ็นเซอร์ เส้นทางเหล่านี้จำเป็นต้องมีการตรวจสอบเป็นประจำ ตามที่ระบุไว้ในคำแนะนำด้านความสมบูรณ์ของข้อมูลของ MHRA และ EU GMP ภาคผนวก 11
สำหรับระบบการผลิตเนื้อสัตว์ที่เพาะเลี้ยง, การควบคุมเหล่านี้มีความสำคัญเป็นพิเศษ เนื่องจากสภาพแวดล้อมมีผลกระทบโดยตรงต่อความมีชีวิตของเซลล์
เมื่อมีการควบคุมเหล่านี้แล้ว ระบบจะต้องได้รับการตรวจสอบความถูกต้องและการเปลี่ยนแปลงจะต้องได้รับการจัดการอย่างรอบคอบเพื่อให้มั่นใจว่ามีการปฏิบัติตามข้อกำหนดอย่างต่อเนื่อง
ขั้นตอนการตรวจสอบและควบคุมการเปลี่ยนแปลง
ระบบการตรวจสอบต้องผ่านการตรวจสอบความถูกต้องผ่านขั้นตอน IQ, OQ, และ PQ เพื่อยืนยันความแม่นยำของเซ็นเซอร์, การทำงานของสัญญาณเตือน, ความสมบูรณ์ของข้อมูล, กระบวนการสำรองข้อมูล, และเส้นทางการตรวจสอบ หรือสามารถใช้วิธีการตามวงจรชีวิตที่สอดคล้องกับหลักการ GAMP 5 และภาคผนวก 11
EU GMP ภาคผนวก 1 (การแก้ไขปี 2022) กำหนดให้ระบบการตรวจสอบสิ่งแวดล้อมต้อง "มีคุณสมบัติและตรวจสอบความถูกต้องอย่างเหมาะสม" และกำหนดให้บันทึกอิเล็กทรอนิกส์ต้องเป็นไปตามมาตรฐานภาคผนวก 11 สำหรับความสมบูรณ์, ความปลอดภัย, และการตรวจสอบย้อนกลับ ข้อกำหนดเหล่านี้เป็นมาตรฐานพื้นฐานสำหรับสถานประกอบการที่สอดคล้องกับ GMP
การเปลี่ยนแปลงใด ๆ ที่อาจส่งผลต่อความสมบูรณ์ของข้อมูล, การทำงานของสัญญาณเตือน, หรือการตรวจสอบย้อนกลับต้องผ่านกระบวนการควบคุมการเปลี่ยนแปลงอย่างเป็นทางการ แม้แต่การอัปเดตที่ดูเหมือนเล็กน้อย เช่น การแก้ไขซอฟต์แวร์ ก็สามารถรบกวนเส้นทางการตรวจสอบได้และไม่ควรดำเนินการโดยไม่มีการประเมินผลกระทบล่วงหน้า
ประเภทข้อมูลที่แตกต่างกันต้องการการควบคุม GMP ที่ปรับให้เหมาะสมเพื่อให้มั่นใจว่าการรายงานมีความถูกต้องและทันเวลา
การเปรียบเทียบประเภทข้อมูลและข้อกำหนดการปฏิบัติตาม
แต่ละประเภทข้อมูลในการตรวจสอบห้องสะอาดมีข้อกำหนดเฉพาะเพื่อรักษาการปฏิบัติตาม GMPตารางด้านล่างนี้สรุปการควบคุมหลักสำหรับประเภทข้อมูลต่างๆ:
| ประเภทข้อมูล | โหมดการตรวจสอบ | การควบคุม GMP หลัก | พื้นฐานขีดจำกัด |
|---|---|---|---|
| การนับอนุภาคที่ไม่สามารถมีชีวิตได้ | ต่อเนื่องหรือบ่อยครั้งระหว่างการดำเนินงานในเกรด A/B; เป็นประจำในเกรดอื่นๆ | เครื่องนับอนุภาคที่ผ่านการตรวจสอบ; การจับข้อมูลอัตโนมัติ; มีสัญญาณเตือน; การสอบเทียบที่สามารถตรวจสอบย้อนกลับไปยังมาตรฐาน; บันทึกการตรวจสอบสำหรับการเปลี่ยนแปลงการกำหนดค่า | ขีดจำกัดคลาส ISO 14644-1; คำแนะนำ Annex 1 สำหรับโซนเกรด A/B |
| ความดันต่าง | ต่อเนื่อง; มีสัญญาณเตือน | เครื่องส่งความดันที่ผ่านการสอบเทียบ; การบันทึกอัตโนมัติพร้อมการประทับเวลา; การยอมรับสัญญาณเตือนที่บันทึกไว้; รักษาความดันต่าง 10–15 Pa ระหว่างโซน | Annex 1; การออกแบบการจำแนกห้องเฉพาะของสถานที่ |
| อุณหภูมิและความชื้นสัมพัทธ์ | ต่อเนื่องสำหรับกระบวนการที่สำคัญ; เป็นระยะในที่อื่น | หัววัดที่ผ่านการสอบเทียบ; การเก็บข้อมูลอัตโนมัติ; การวิเคราะห์แนวโน้ม; ขีดจำกัดการเตือนตามความต้องการของกระบวนการและข้อกำหนดทางกฎหมาย | ความรู้เกี่ยวกับกระบวนการ; คำแนะนำทางกฎหมาย; ความไวของผลิตภัณฑ์ |
| จุลินทรีย์ในอากาศที่มีชีวิต | เป็นระยะ (การเก็บตัวอย่างอากาศแบบแอคทีฟ); ความถี่ที่เพิ่มขึ้นสำหรับการดำเนินงานที่สำคัญ | ผู้เก็บตัวอย่างที่ผ่านการรับรอง; ขั้นตอนการเก็บตัวอย่างที่ควบคุม; การส่งต่อไปยังห้องปฏิบัติการ; ผลลัพธ์เชื่อมโยงกับชุดและสถานที่; บันทึกพร้อมสำหรับการตรวจสอบ | ขีดจำกัดจุลินทรีย์ตามเกรดของ EU GMP ภาคผนวก 1 |
| ผลการสัมผัสพื้นผิว | เป็นระยะ; หลังการทำความสะอาดและหลังการดำเนินงาน | วิธีการสุ่มตัวอย่างที่ควบคุม; การตรวจสอบย้อนกลับในห้องปฏิบัติการ; ผลลัพธ์ที่ตรวจสอบเทียบกับขีดจำกัดเฉพาะเกรด; เชื่อมโยงกับบันทึกการทำความสะอาด | EU GMP Annex 1; SOP ของสถานที่ |
ข้อมูลแต่ละประเภทต้องการเกณฑ์การยอมรับที่กำหนดไว้, ตารางการตรวจสอบปกติ, นโยบายการเก็บรักษา, และกระบวนการยกระดับสำหรับการเบี่ยงเบน การใช้มาตรฐานการตรวจสอบที่เหมือนกันกับข้อมูลทุกประเภทเป็นข้อผิดพลาดทั่วไปที่หน่วยงานกำกับดูแลกำลังตรวจสอบอย่างเข้มงวดมากขึ้น การปรับกระบวนการตรวจสอบให้เหมาะสมกับความต้องการเฉพาะของแต่ละประเภทข้อมูลช่วยให้มั่นใจได้ถึงการปฏิบัติตามข้อกำหนดและประสิทธิภาพในการดำเนินงาน
การรายงาน, การตรวจสอบ, และการดำเนินการแก้ไข
การสร้างรายงานที่สอดคล้องกับข้อกำหนด
รายงานที่สอดคล้องกับมาตรฐาน ALCOA+ ต้องมีความละเอียด, แม่นยำ, และสามารถเข้าถึงได้สำหรับการตรวจสอบ รายงานการตรวจสอบห้องสะอาดที่สอดคล้องกับ GMP ควรมีความกระชับ, ตรวจสอบได้, และสามารถสนับสนุนการตัดสินใจในการปล่อยชุดผลิตภัณฑ์ในขณะที่แสดงการควบคุมสิ่งแวดล้อม อย่างน้อยที่สุด รายงานเหล่านี้ควร:
- ครอบคลุมช่วงเวลาและขอบเขตการตรวจสอบ
- สรุปกิจกรรมการสุ่มตัวอย่างเมื่อเปรียบเทียบกับตารางที่วางแผนไว้
- ระบุอย่างชัดเจนว่ามีการเกินขีดจำกัดการแจ้งเตือนหรือการดำเนินการหรือไม่
การวิเคราะห์แนวโน้มเป็นส่วนสำคัญของรายงานเหล่านี้ โดยใช้เครื่องมือทางสถิติเช่น แผนภูมิควบคุม ค่าเฉลี่ยเคลื่อนที่ และอัตราการเบี่ยงเบนต่อ 100 ตัวอย่าง เพื่อระบุการเปลี่ยนแปลงที่ค่อยเป็นค่อยไป ตัวอย่างเช่น แนวโน้มรายเดือนที่แสดงการเพิ่มขึ้นอย่างต่อเนื่องของจำนวนที่สามารถดำรงชีวิตได้ใกล้กับสายการเก็บเกี่ยวของเครื่องปฏิกรณ์ชีวภาพให้ข้อมูลเชิงลึกมากกว่าการเกิดเหตุการณ์ที่เกินขีดจำกัดเพียงครั้งเดียว การเพิ่มคำอธิบายประกอบ เช่น กิจกรรมการบำรุงรักษา การปรับกระบวนการ หรือการเปลี่ยนแปลงบุคลากร ทำให้ข้อมูลตีความได้ง่ายขึ้นและพร้อมสำหรับการตรวจสอบมากขึ้น
การตรวจสอบเส้นทางการตรวจสอบเป็นอีกขั้นตอนสำคัญที่ต้องการบุคลากรที่ผ่านการฝึกอบรมเพื่อบันทึกผลการค้นพบอย่างละเอียด ซึ่งรวมถึงการบันทึกว่าใครเป็นผู้ตรวจสอบเหตุการณ์ระบบเฉพาะ การบันทึกความผิดปกติใด ๆ และการระบุการดำเนินการติดตามผล ทั้งหมดนี้อยู่ในบันทึกที่ลงนามและลงวันที่
ความถี่ของรายงานควรสอดคล้องกับความเสี่ยงที่เกี่ยวข้อง ตัวอย่างเช่น:
- รายงานที่เกี่ยวข้องกับชุดการผลิตจะถูกสร้างขึ้นสำหรับการผลิตแต่ละครั้ง
- สรุปการตรวจสอบสิ่งแวดล้อมตามปกติมักจะเป็นรายสัปดาห์หรือรายเดือน.
- รายงานแนวโน้มจะถูกจัดทำขึ้นเป็นรายเดือนหรือรายไตรมาสเพื่อระบุสัญญาณเริ่มต้นของการเปลี่ยนแปลง.
ความถี่ในการรายงานที่เลือกต้องได้รับการอธิบายในขั้นตอนการปฏิบัติงานมาตรฐาน (SOPs) และปฏิบัติตามอย่างสม่ำเสมอ โปรโตคอลเหล่านี้ยังเป็นพื้นฐานสำหรับการเริ่มต้นการดำเนินการแก้ไขเมื่อพบการเบี่ยงเบน.
การจัดการความล้มเหลวในการปฏิบัติตามข้อกำหนด
เมื่อเกิดการเบี่ยงเบน การตอบสนองที่มีโครงสร้างและสามารถติดตามได้เป็นสิ่งสำคัญ แต่ละการเบี่ยงเบนควรมีตัวระบุเฉพาะ คำอธิบายที่ชัดเจน และการประเมินความเสี่ยงที่ประเมินทั้งผลกระทบต่อผลิตภัณฑ์และความสมบูรณ์ของข้อมูล การเบี่ยงเบนต้องถูกจัดประเภท (เล็กน้อย, สำคัญ, หรือวิกฤติ) และเชื่อมโยงกับชุดหรือการผลิตเฉพาะเพื่อประเมินว่าการปล่อยชุดได้รับผลกระทบหรือจำเป็นต้องมีการทดสอบเพิ่มเติมหรือไม่.
กรอบงาน CAPA (การดำเนินการแก้ไขและป้องกัน) เป็นศูนย์กลางในการจัดการกับความล้มเหลวของ GMP การดำเนินการ CAPA อย่างมีประสิทธิภาพต้องการมากกว่าการระบุเหตุการณ์ว่าเป็น "ความผิดพลาดของมนุษย์" คำแนะนำของ EMA และ PIC/S เน้นย้ำ:
"ความล้มเหลวในการสืบสวนการเบี่ยงเบนที่สำคัญ, ผลลัพธ์ OOS และปัญหาความสมบูรณ์ของข้อมูลอย่างเพียงพอ" เป็นสาเหตุที่เกิดขึ้นซ้ำๆ ของการดำเนินการบังคับใช้.
เครื่องมือวิเคราะห์สาเหตุรากฐาน เช่น 5 Whys หรือแผนภูมิก้างปลา มีคุณค่าอย่างยิ่งในการค้นหาปัญหาระบบ - ไม่ว่าจะเกี่ยวข้องกับช่องว่างในกระบวนการ, การฝึกอบรมที่ไม่เพียงพอ, หรือจุดอ่อนในการควบคุมทางเทคนิค การดำเนินการแก้ไขควรจัดการกับความเสี่ยงที่ระบุในระหว่างการจับและจัดเก็บข้อมูล.
แต่ละ CAPA ต้องมีเกณฑ์ประสิทธิผลที่สามารถวัดได้ ตัวอย่างเช่น: "ไม่มีการเบี่ยงเบนของ Grade B ที่มีชีวิตซ้ำซ้อนเกินระดับการดำเนินการเป็นเวลาหกเดือน" นอกจากนี้ การทบทวนติดตามผลเป็นสิ่งสำคัญเพื่อให้แน่ใจว่าเกณฑ์เหล่านี้ได้รับการปฏิบัติ. ตัวชี้วัด CAPA ทั่วไปประกอบด้วย:
- จำนวนของการดำเนินการที่ยังเปิดอยู่.
- เวลาเฉลี่ยในการปิด.
- เปอร์เซ็นต์ของการดำเนินการที่เสร็จสิ้นตรงเวลา.
- อัตราการเบี่ยงเบนซ้ำ ซึ่งเป็นตัวบ่งชี้ที่แข็งแกร่งของประสิทธิภาพ CAPA.
การทบทวนจดหมายเตือน GMP จากปี 2015 ถึง 2019 พบว่า 65–70% ของการอ้างอิงความสมบูรณ์ของข้อมูลเกิดจากการสอบสวนที่ไม่เพียงพอ การขาดเอกสาร หรือความล้มเหลวในการตรวจสอบและรายงานข้อมูลอย่างถูกต้อง [2]. สิ่งนี้เน้นย้ำถึงความสำคัญของการรายงานที่แข็งแกร่งและกรอบงาน CAPA ที่ตอบสนองได้เป็นหลักฐานของสถานที่ที่มีการควบคุมอย่างดี.
การรักษาความสอดคล้องของ GMP ในสถานที่ผลิตเนื้อสัตว์ที่เพาะเลี้ยง
เพื่อให้มั่นใจในความปลอดภัยและคุณภาพในการผลิตเนื้อสัตว์ที่เพาะเลี้ยง สถานที่ต้องปรับการควบคุม GMP ที่มีอยู่ให้สอดคล้องกับความท้าทายเฉพาะของสาขาใหม่นี้. เนื่องจากความสมบูรณ์ของข้อมูลในห้องปลอดเชื้อมีบทบาทสำคัญในการรักษาความปลอดภัยของผลิตภัณฑ์ การปรับปรุงแนวปฏิบัติ GMP สำหรับเนื้อสัตว์ที่เพาะเลี้ยงจึงเป็นสิ่งจำเป็น
การปรับแต่งแนวปฏิบัติ GMP สำหรับเนื้อสัตว์ที่เพาะเลี้ยง
กรอบงาน GMP เช่น EU Annex 1 ซึ่งเดิมสร้างขึ้นสำหรับเภสัชภัณฑ์ จำเป็นต้องมีการปรับเปลี่ยนเพื่อจัดการกับความเสี่ยงเฉพาะในกระบวนการผลิตเนื้อสัตว์ที่เพาะเลี้ยง การประเมินความเสี่ยงอย่างเป็นทางการ เช่น การวิเคราะห์แบบ FMEA หรือ HACCP ให้พื้นฐานที่มั่นคงสำหรับการปรับแนวปฏิบัติ GMP ให้สอดคล้องกับแต่ละขั้นตอนของการผลิต การดำเนินงานที่สำคัญ เช่น การละลายเซลล์แบงค์ การฉีดเชื้อในเครื่องปฏิกรณ์ชีวภาพ การขยายเซลล์ และการเก็บเกี่ยว ต้องการการจัดประเภทห้องปลอดเชื้อที่เหมาะสม โปรโตคอลการสวมใส่เสื้อผ้า และการตรวจสอบสิ่งแวดล้อมตามที่ระบุใน Annex 1ในขณะเดียวกัน งานที่ต้องทำต่อเนื่องเช่น การจัดการโครงสร้างและการบรรจุภัณฑ์สามารถปฏิบัติตามมาตรฐาน GMP ด้านสุขอนามัยระดับอาหารภายใต้ Regulation (EC) No 852/2004, โดยที่การติดตามและความสมบูรณ์ของข้อมูลยังคงอยู่ตลอดกระบวนการ [6] [9][14].
กลยุทธ์การตรวจสอบสิ่งแวดล้อมควรมุ่งเน้นไปที่สิ่งมีชีวิตที่เกี่ยวข้องกับเนื้อสัตว์ที่เพาะเลี้ยงและความปลอดภัยของอาหาร แทนที่จะมุ่งเป้าไปที่เชื้อโรคทางเภสัชกรรมแบบดั้งเดิม การสุ่มตัวอย่างควรให้ความสำคัญในพื้นที่ที่มีความเสี่ยงสูง เช่น บริเวณใกล้เครื่องปฏิกรณ์ชีวภาพที่เปิดอยู่ โซนเตรียมสื่อ และสถานีจัดการโครงสร้าง สถานที่เหล่านี้ควรเลือกตามรูปแบบการไหลของอากาศที่บันทึกไว้และการวิเคราะห์การเคลื่อนไหวของบุคลากร [9][10].
เนื่องจากปริมาณข้อมูลที่สูงที่เกิดจากเครื่องปฏิกรณ์ชีวภาพเนื้อสัตว์เพาะเลี้ยง ระบบจะต้องสามารถจับภาพ ประทับเวลา และจัดเก็บข้อมูลนี้อย่างปลอดภัยในที่เก็บข้อมูลที่ผ่านการตรวจสอบแล้ว ไฟล์ข้อมูลดิบต้นฉบับจากเครื่องมือควรได้รับการระบุเสมอว่าเป็นบันทึกหลักเพื่อให้แน่ใจว่าปฏิบัติตาม [7][8].
โปรโตคอลการทำความสะอาดและการฆ่าเชื้อยังต้องการการพิจารณาอย่างรอบคอบ คราบตกค้างที่ถือว่ายอมรับได้ในสภาพแวดล้อมทางเภสัชกรรมอาจรบกวนการยึดเกาะหรือการแยกแยะเซลล์ในการผลิตเนื้อสัตว์เพาะเลี้ยง ข้อมูลการตรวจสอบสำหรับสารทำความสะอาดควรถูกรวบรวมและเก็บรักษาไว้เป็นส่วนหนึ่งของโปรแกรมการตรวจสอบสิ่งแวดล้อม [3][4].
การใช้ทรัพยากรในอุตสาหกรรม เช่น Cellbase
แพลตฟอร์มการจัดซื้อเฉพาะทางมีคุณค่าอย่างยิ่งในการตอบสนองความต้องการเฉพาะของโรงงานผลิตเนื้อสัตว์เพาะเลี้ยง อุปกรณ์ห้องสะอาดที่เข้ากันได้กับ GMP ควรเป็นไปตามมาตรฐานการป้องกันการรั่วไหลและการทำความสะอาด ในขณะเดียวกันก็ควรรวมเข้ากับระบบข้อมูลที่ผ่านการตรวจสอบแล้วอย่างไร้รอยต่อ ซัพพลายเออร์ต้องให้รายละเอียดข้อมูลจำเพาะ รวมถึงความสามารถในการติดตามการตรวจสอบ รูปแบบการส่งออกข้อมูล การกำหนดค่าการเตือนภัย และขั้นตอนการสอบเทียบ ควบคู่ไปกับอุปกรณ์
เมื่อจัดหาระบบผ่าน
- คุณสมบัติความสมบูรณ์ของข้อมูล: เส้นทางการตรวจสอบที่ปลอดภัย, บันทึกที่มีการประทับเวลา, การอนุญาตตามบทบาท, และการเข้าสู่ระบบของผู้ใช้ที่ไม่ซ้ำกัน
- ความเข้ากันได้ของระบบ: การสนับสนุนโปรโตคอลการสื่อสารมาตรฐานและ API สำหรับการจัดเก็บข้อมูลแบบรวมศูนย์
- การสอบเทียบและการบำรุงรักษา: การมีเอกสารประกอบที่ครอบคลุม
- การสนับสนุนการรับรองคุณภาพ: แม่แบบ IQ/OQ ที่ผู้จัดหาจัดเตรียมไว้สำหรับการตรวจสอบที่มีประสิทธิภาพ
- ความเหมาะสมของห้องสะอาด: วัสดุและการออกแบบที่เอื้อต่อการทำความสะอาด
การขอเอกสารจากผู้จัดหาตั้งแต่เนิ่นๆ ในกระบวนการจัดซื้อสามารถช่วยหลีกเลี่ยงปัญหาการรับรองคุณภาพที่อาจเกิดขึ้นในภายหลัง [3][4].
บทสรุป
สรุปประเด็นสำคัญ
การปฏิบัติตาม GMP ในการจัดการข้อมูลห้องสะอาดเกี่ยวข้องกับการแสดงการควบคุม - ทั้งในกระบวนการ บันทึก และการตัดสินใจที่ได้รับข้อมูลจากข้อมูลนั้น หากบันทึกขาดความน่าเชื่อถือ กระบวนการที่บันทึกไว้ก็จะกลายเป็นที่น่าสงสัยเช่นกัน หลักการนี้ใช้ได้ทั่วไป ไม่ว่าจะเป็นการจัดการบันทึกการตรวจสอบสิ่งแวดล้อม ผลลัพธ์จากเครื่องปฏิกรณ์ชีวภาพ รายงานการเบี่ยงเบน หรือใบรับรองการสอบเทียบ
มีสี่หัวข้อหลักที่เกิดขึ้นตลอดการสนทนานี้ ประการแรก ความสมบูรณ์ของข้อมูล, ที่นำโดยหลักการ ALCOA+ เป็นรากฐานของเอกสารห้องสะอาดที่สอดคล้องกัน ประการที่สอง การจัดการวงจรชีวิต ช่วยให้มั่นใจว่าข้อมูลถูกบันทึกอย่างถูกต้อง ตรวจสอบอย่างรวดเร็ว เก็บไว้อย่างปลอดภัย และรักษาไว้ตามระยะเวลาที่กำหนด ประการที่สาม ระบบการตรวจสอบที่ผ่านการตรวจสอบและควบคุมการเปลี่ยนแปลง เป็นรากฐานทางเทคนิคที่ไม่มี SOP ใดสามารถทดแทนได้ตามที่เน้นโดยการวิเคราะห์ของ MHRA เกี่ยวกับการตรวจสอบ GMP ตั้งแต่ปี 2016 ถึง 2021 ข้อบกพร่องทั่วไปยังคงรวมถึงบันทึกที่ไม่สมบูรณ์และการตรวจสอบเส้นทางการตรวจสอบที่ไม่เพียงพอ [1]. สุดท้าย การรายงานที่ถูกต้องและสามารถตรวจสอบได้ ช่วยให้ข้อมูลดิบสามารถเชื่อมโยงกับการตัดสินใจเกี่ยวกับชุดการผลิต การสืบสวน และการดำเนินการแก้ไข ซึ่งตรงตามความคาดหวังของกฎระเบียบ
สำหรับโรงงานผลิตเนื้อสัตว์ที่เพาะเลี้ยง หลักการเหล่านี้มีความสำคัญมากยิ่งขึ้น ความท้าทายในการรวมเวิร์กโฟลว์แบบ R&D เข้ากับการควบคุมระดับการผลิตต้องการการจัดการข้อมูลที่แข็งแกร่งเพื่อเชื่อมโยงสภาพแวดล้อมการดำเนินงานทั้งสอง การจัดการข้อมูลในห้องสะอาดอย่างเหมาะสมไม่เพียงแต่รับประกันความสม่ำเสมอและการทำซ้ำได้ แต่ยังเตรียมโรงงานสำหรับการขยายขนาดในขณะที่แสดงความปลอดภัยของผลิตภัณฑ์ต่อหน่วยงานกำกับดูแล นักลงทุน และผู้บริโภค
คำแนะนำที่สามารถนำไปปฏิบัติได้มากที่สุด? จัดการความเสี่ยงที่ทราบก่อนที่ผู้ตรวจสอบจะเน้นย้ำช่องโหว่เช่นระบบกระดาษ-อิเล็กทรอนิกส์แบบผสม, การแชร์การเข้าสู่ระบบของผู้ใช้, การตรวจสอบข้อมูลที่ล่าช้า, และการจัดเก็บข้อมูลในท้องถิ่นที่ไม่สามารถควบคุมได้สามารถคาดการณ์และป้องกันได้ การแก้ไขปัญหาเหล่านี้อย่างเชิงรุกมีประสิทธิภาพมากกว่า - และมีค่าใช้จ่ายน้อยกว่า - การสร้างเส้นทางข้อมูลใหม่หลังจากเกิดเหตุการณ์คุณภาพ
สำหรับทีมที่กำลังมองหาอุปกรณ์ตรวจสอบ, เซ็นเซอร์, หรือโครงสร้างพื้นฐานที่ปรับให้เหมาะสมกับความต้องการเหล่านี้,
ขั้นตอนเหล่านี้มีความสำคัญต่อการปฏิบัติตาม Good Manufacturing Practice (GMP) ในการจัดการข้อมูลในห้องสะอาด.
ความเสี่ยงหลักของความสมบูรณ์ของข้อมูลในระบบกระดาษ-อิเล็กทรอนิกส์แบบผสมคืออะไร?
การจัดเก็บข้อมูลในหลายสถานที่ทำให้เกิดความซับซ้อนในการตรวจสอบความถูกต้องของข้อมูล นอกจากนี้ การป้อนข้อมูลด้วยตนเองยังเพิ่มความเสี่ยงของข้อผิดพลาดจากมนุษย์ ในขณะที่ระบบที่ควบคุมไม่ดีหรือระบบที่แยกออกจากกันทำให้บันทึกมีความเสี่ยงต่อการถูกแก้ไขหรือการลบ ปัญหาเหล่านี้เน้นย้ำถึงความจำเป็นในการจัดการข้อมูลที่แข็งแกร่งเพื่อรักษาการปฏิบัติตามข้อกำหนดและรักษาความสมบูรณ์ของข้อมูล
หลักฐานอะไรที่ผู้ตรวจสอบคาดหวังสำหรับการตรวจสอบความถูกต้องของระบบและการควบคุมการเปลี่ยนแปลง?
ผู้ตรวจสอบมักจะขอ หลักฐานที่เป็นเอกสาร ที่แสดงถึงการตรวจสอบความถูกต้องของระบบ ซึ่งรวมถึงการทดสอบพารามิเตอร์ที่สำคัญ เช่น:
- ความสมบูรณ์ของตัวกรอง HEPA: การรับรองว่าตัวกรองตรงตามมาตรฐานประสิทธิภาพที่กำหนด
- การไหลของอากาศและความแตกต่างของความดัน: การตรวจสอบว่าค่าเหล่านี้อยู่ในช่วงที่ยอมรับได้เพื่อรักษาสภาพแวดล้อมที่ควบคุมได้
- ข้อมูลการตรวจสอบสิ่งแวดล้อม: แสดงให้เห็นว่าสถานที่นั้นเป็นไปตามข้อกำหนดด้านความสะอาดและการควบคุมการปนเปื้อน
นอกเหนือจากการทดสอบการตรวจสอบความถูกต้อง การบันทึกกิจกรรมการควบคุมการเปลี่ยนแปลงก็มีความสำคัญเช่นกัน ซึ่งครอบคลุมถึงการกระทำเช่นการเปลี่ยนไส้กรองหรือการปรับปรุงสถานที่, ซึ่งช่วยพิสูจน์ว่าระบบยังคงทำงานตามที่คาดหวังและเป็นไปตามมาตรฐานข้อบังคับ










