

บันทึกการผลิตมีความสำคัญอย่างยิ่งต่อการปฏิบัติตามข้อกำหนดและความปลอดภัยของผลิตภัณฑ์ พวกเขาบันทึกทุกขั้นตอนของการผลิตเพื่อให้มั่นใจว่ามาตรฐานการกำกับดูแลได้รับการปฏิบัติตาม สำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง การรักษาความปลอดเชื้อและบันทึกที่ละเอียดเป็นสิ่งที่ไม่สามารถต่อรองได้ การตรวจสอบของ FDA มักจะเน้นปัญหาเช่นข้อมูลที่ขาดหายไป การตรวจสอบที่ไม่สมบูรณ์ และการดำเนินการแก้ไขที่ไม่ดี ซึ่งอาจนำไปสู่การเตือนหรือการหยุดชะงัก
ประเด็นสำคัญ:
- บันทึกการผลิต: มีสองประเภท - บันทึกการผลิตหลัก (MBR) (คือ "สูตร") และบันทึกการผลิตแบทช์ (BPR) (คือ "การดำเนินการ")
- ปัญหาทั่วไป: ข้อผิดพลาดของมนุษย์ (50% ของปัญหา) การตรวจสอบระหว่างกระบวนการที่ขาดหายไป , การตรวจสอบที่ไม่สมบูรณ์ และระบบ CAPA (การแก้ไขและป้องกัน) ที่ไม่ดี
- มาตรฐานของ FDA: การปฏิบัติตามหลักการ ALCOA+ (สามารถระบุได้ อ่านออก เขียนได้ทันที ต้นฉบับ ถูกต้อง สมบูรณ์ สม่ำเสมอ ทนทาน พร้อมใช้งาน) เป็นสิ่งจำเป็น
- โซลูชัน: การตรวจสอบอิสระ, บันทึกชุดอิเล็กทรอนิกส์, และการตรวจสอบซัพพลายเออร์อย่างเข้มงวดสามารถลดข้อผิดพลาดและปรับปรุงการปฏิบัติตามข้อกำหนดได้.
บริษัทเนื้อสัตว์เพาะเลี้ยงเช่น UPSIDE Foods ได้ตั้งมาตรฐานโดยการรับรองเอกสารที่ละเอียด, การติดตามแหล่งที่มา, และการแก้ไขปัญหาอย่างรวดเร็ว. โดยการเรียนรู้จากแนวปฏิบัติเหล่านี้, ผู้ผลิตสามารถหลีกเลี่ยงปัญหาด้านกฎระเบียบและรักษามาตรฐานคุณภาพสูง.
คู่มือที่ครอบคลุมเกี่ยวกับการจัดทำเอกสารและการเก็บบันทึกสำหรับ FDA การปฏิบัติตามข้อกำหนดในวิทยาศาสตร์ชีวภาพ
sbb-itb-ffee270
ปัญหาทั่วไปในการจัดทำบันทึกชุดการผลิต
รายงานการตรวจสอบของ FDA มักเน้นปัญหาที่เกิดขึ้นซ้ำ: การเบี่ยงเบนในการตรวจสอบบันทึกการผลิตเป็นหนึ่งในข้อบกพร่อง GMP ที่ถูกอ้างถึงมากที่สุดโดยหน่วยงานกำกับดูแล [7]. สำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง ข้อบกพร่องเหล่านี้เกินกว่าความผิดพลาดทางการบริหาร - พวกเขาเสี่ยงต่อความสามารถในการแสดงสภาพปลอดเชื้อที่ยั่งยืน ปัญหาเหล่านี้ปรากฏในหลายรูปแบบ ดังที่แสดงในตัวอย่างด้านล่างนี้
การตรวจสอบที่ไม่สมบูรณ์และการไม่ปฏิบัติตามข้อกำหนด
ปัญหาทั่วไปอย่างหนึ่งคือการที่หน่วยควบคุมคุณภาพไม่สามารถตรวจสอบบันทึกชุดการผลิตได้อย่างละเอียด แทนที่จะเป็นส่วนสำคัญของกระบวนการปล่อยผลิตภัณฑ์ การตรวจสอบมักเกิดขึ้นแบบตอบสนอง - เฉพาะเมื่อปัญหาผลิตภัณฑ์ได้เกิดขึ้นแล้ว [7]. วิธีการนี้ทำให้เกิดช่องว่างที่สำคัญในบันทึกการผลิต
ตัวอย่างเช่น Davis City Pharmacy ได้รับการสังเกตจาก FDA 483 เนื่องจากบันทึกชุดการผลิตขาดรายละเอียดสำคัญ เช่น ปริมาณส่วนประกอบ ขั้นตอนการดำเนินงาน และลายเซ็นของบุคลากร ในทำนองเดียวกัน CAPS ถูกอ้างถึงเนื่องจากขาดลายเซ็นที่จำเป็นและการตรวจสอบของผู้ตรวจสอบในรายการสำคัญ [3]. เหตุการณ์เหล่านี้ไม่ใช่เหตุการณ์ที่เกิดขึ้นเพียงครั้งเดียว; การศึกษาพบว่าประมาณ 52% ของการละเมิดเอกสารจะเพิ่มขึ้นเมื่อไม่มี ระบบการจัดการกระบวนการชีวภาพ ที่มีประสิทธิภาพ [3].
"ไม่ใช่ความซับซ้อนของกระบวนการที่ทำให้เกิดการอ้างอิง - แต่เป็นความไม่สม่ำเสมอ, ความไม่สมบูรณ์, และการควบคุมที่ไม่ดี." - GXP Auditing & Consulting Services [5]
บันทึกการตรวจสอบระหว่างกระบวนการที่ขาดหายไป
ข้อบกพร่องที่พบบ่อยอีกประการหนึ่งคือการขาดเอกสารที่เหมาะสมสำหรับการตรวจสอบระหว่างกระบวนการ บันทึกเหล่านี้มีความสำคัญโดยเฉพาะอย่างยิ่งในจุดควบคุมที่สำคัญในกระบวนการปลอดเชื้อ ตัวอย่างเช่น Nephron Sterile Compounding Center ถูกอ้างอิงเนื่องจากไม่สามารถบันทึกขั้นตอนการสวมใส่เสื้อผ้าที่จำเป็นและขั้นตอนปลอดเชื้อในบันทึกชุดของพวกเขา [3]. สำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง ซึ่งความสะอาดเป็นสิ่งสำคัญ การละเว้นดังกล่าวทำให้ไม่สามารถยืนยันการปฏิบัติตาม มาตรการควบคุมการปนเปื้อน.
Amphastar ยังถูกระบุว่าไม่สามารถตรวจสอบหรือบันทึกความแปรปรวนของผลผลิตที่ไม่คาดคิดหรือความคลาดเคลื่อนของผลลัพธ์ [3]. ความเสี่ยงของการละเลยดังกล่าวมีความชัดเจน ในกรณีหนึ่ง โรงงานผลิตยาที่ไม่ระบุชื่อในปี 2024/2025 ถูกพบว่ามีการเก็บบันทึกชุดงานที่เสร็จสมบูรณ์บนชั้นวางและโต๊ะทำงานที่เปิดโล่ง ผู้ตรวจสอบของ FDA พบหน้าที่หายไป รวมถึงเจ็ดหน้าจากบันทึกเดียว และส่วน "Synthesis Solution" ทั้งหมดหายไปจากอีกบันทึกหนึ่ง [6].
ความล้มเหลวของ CAPA และการตรวจสอบ GMP ของผู้จัดหา
นอกเหนือจากข้อผิดพลาดในการบันทึกเอกสาร การขาดกระบวนการแก้ไขที่มีประสิทธิภาพและการตรวจสอบผู้จัดหายังบั่นทอนความน่าเชื่อถือของบันทึกชุดงานอีกด้วยเมื่อเกิดการเบี่ยงเบนในการผลิตโดยไม่มีรายงานการแก้ไขและป้องกัน (CAPA) ที่สอดคล้องกัน ความสมบูรณ์ของบันทึกชุดการผลิตจะถูกทำลาย [7]. ตัวอย่างเช่น Eugia Pharma Specialities Limited, ที่ได้รับการตรวจสอบระหว่างวันที่ 22 มกราคม ถึง 2 กุมภาพันธ์ 2024 ได้รับ FDA 483 เนื่องจากไม่สามารถตรวจสอบความคลาดเคลื่อนได้อย่างเพียงพอ ระบบ CAPA ที่ไม่มีประสิทธิภาพและการสืบสวนที่ไม่สมบูรณ์ของพวกเขานำไปสู่ปัญหาการผลิตซ้ำซาก ทำให้ต้องปรับปรุงกระบวนการสืบสวนและ CAPA ของพวกเขาใหม่ทั้งหมด [9].
ในทำนองเดียวกัน ในระหว่างการตรวจสอบตั้งแต่วันที่ 26 กันยายน ถึง 25 ตุลาคม 2023 Stokes Healthcare Inc. แสดงให้เห็นถึงการจัดการความคลาดเคลื่อนที่ไม่ดี บริษัทไม่สามารถขยายการสืบสวนไปยังชุดที่ได้รับผลกระทบทั้งหมดและล่าช้าในการทำการวิเคราะห์ให้เสร็จสมบูรณ์ [9].
"ความคลาดเคลื่อนที่ไม่มีรายงาน CAPA หรือการเบี่ยงเบนที่สอดคล้องกัน? นั่นคือความล้มเหลวในการปฏิบัติตามข้อกำหนด" - GXP Auditing & Consulting Services [5]
ปัญหาการตรวจสอบซัพพลายเออร์เพิ่มความซับซ้อนอีกชั้นหนึ่ง Empower Clinic Services LLC ถูกอ้างถึงระหว่างการตรวจสอบตั้งแต่วันที่ 18 กรกฎาคม ถึง 5 สิงหาคม 2022 เนื่องจากขั้นตอนการควบคุมคุณภาพที่ไม่เพียงพอ รวมถึงคุณสมบัติของซัพพลายเออร์ที่ไม่เพียงพอและกระบวนการตรวจสอบที่ไม่ดี [9]. สำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง ซึ่งพึ่งพาสื่อการเจริญเติบโต สายเซลล์ และปัจจัยสำคัญอื่น ๆ การรับรองการปฏิบัติตาม GMP ของซัพพลายเออร์เป็นสิ่งสำคัญในการรักษาความสมบูรณ์ของบันทึกชุดผลิตภัณฑ์
ข้อกำหนดของ FDA สำหรับบันทึกชุดผลิตภัณฑ์
กฎของ FDA สำหรับบันทึกชุดผลิตภัณฑ์หมุนรอบ 21 CFR Part 117, ซึ่งกำหนดมาตรฐานพื้นฐานสำหรับความปลอดภัยของอาหารเมื่อพูดถึงเนื้อสัตว์ที่เพาะเลี้ยง ซึ่งการรักษาความปลอดเชื้อในระยะการเพาะเลี้ยงเซลล์เป็นสิ่งสำคัญ เอกสารมักจะต้องเป็นไปตามมาตรฐานที่เข้มงวดกว่า Part 111 หรือ Part 211 , นอกเหนือจาก Part 117 [10][14]. สิ่งนี้เน้นย้ำว่าเอกสารที่แม่นยำเป็นสิ่งสำคัญสำหรับการรับรองความปลอดภัยและประสิทธิภาพของการผลิตเนื้อสัตว์ที่เพาะเลี้ยง
มาตรฐานหลักสำหรับบันทึกการผลิต
แต่ละชุดต้องการเอกสารสำคัญสองฉบับ:
- บันทึกการผลิตหลัก (MBR): แม่แบบที่ได้รับการอนุมัติซึ่งระบุขั้นตอนการผลิต
- บันทึกการผลิตชุด (BPR): บันทึกรายละเอียดของสิ่งที่เกิดขึ้นจริงระหว่างการผลิต [12][2].
BPR ต้องรวมรายละเอียดเฉพาะเช่น หมายเลขแบทช์หรือล็อต รายละเอียดอุปกรณ์ วันที่ทำความสะอาด ตัวระบุส่วนประกอบ การวัดที่แน่นอน และการเปรียบเทียบระหว่างผลผลิตจริงกับผลผลิตตามทฤษฎี [10][14].
"บันทึกการผลิตแบทช์ต้องปฏิบัติตามบันทึกการผลิตหลักที่เหมาะสมอย่างถูกต้อง และคุณต้องดำเนินการแต่ละขั้นตอนในการผลิตแบทช์" – 21 CFR 111.255 [12]
ทุกขั้นตอนสำคัญต้องถูกบันทึกทันที พร้อมทั้งมีการลงชื่อย่อของผู้ดำเนินการและผู้ตรวจสอบ [10][11]. FDA กำหนดให้ปฏิบัติตามหลักการ ALCOA(+) ซึ่งหมายถึงบันทึกต้อง สามารถระบุได้ ชัดเจน ทันเวลา ต้นฉบับ และถูกต้อง - รวมถึง ครบถ้วน สม่ำเสมอ ทนทาน และพร้อมใช้งาน [1] .
หากมีการเบี่ยงเบนจากบันทึกการผลิตหลัก จะต้องมีการตรวจสอบอย่างละเอียด ซึ่งรวมถึงการบันทึกปัญหา การวิเคราะห์สาเหตุที่แท้จริง และการดำเนินการตามแผน Corrective and Preventive Action (CAPA) [8] [1]. การประเมินเบื้องต้นของการเบี่ยงเบนควรถูกบันทึกภายใน 24–48 ชั่วโมงหลังจากการตรวจพบ [8]. สำหรับสถานที่ที่ใช้ระบบอิเล็กทรอนิกส์ การปฏิบัติตาม 21 CFR Part 11 เป็นสิ่งจำเป็น ซึ่งรวมถึงลายเซ็นอิเล็กทรอนิกส์ที่ผ่านการตรวจสอบและเส้นทางการตรวจสอบที่ปลอดภัยและมีการประทับเวลา [8] [1].
ขั้นตอนการเก็บรักษาและตรวจสอบบันทึก
กระบวนการเก็บรักษาและตรวจสอบบันทึกที่เหมาะสมมีความสำคัญต่อการปฏิบัติตามข้อกำหนดและการรับรองความปลอดภัยของผลิตภัณฑ์ในการผลิตที่ปลอดเชื้อ เช่น การผลิตเนื้อสัตว์ที่เพาะเลี้ยง รายละเอียดทุกอย่างในบันทึกการผลิตต้องได้รับการตรวจสอบอย่างละเอียด ทีม ควบคุมคุณภาพ (QC) มีหน้าที่ในการตรวจสอบบันทึกการผลิตทั้งหมด ตรวจสอบผลลัพธ์ และข้อมูลการทดสอบก่อนที่ชุดการผลิตจะได้รับการอนุมัติให้จัดจำหน่าย [10] [13].
"บันทึกการผลิตและการควบคุมผลิตภัณฑ์ยาทั้งหมดจะต้องได้รับการตรวจสอบและอนุมัติโดยหน่วยควบคุมคุณภาพก่อนที่ชุดการผลิตจะถูกปล่อยหรือจัดจำหน่าย" – 21 CFR 211.192 [2]
ผู้ผลิตมักตั้งเป้าที่จะทำการตรวจสอบ 95% ของบันทึกการผลิตภายใน 30 วัน หลังการผลิต [2] . อย่างไรก็ตาม สำหรับกระบวนการปลอดเชื้อที่ซับซ้อนมากขึ้นที่เกี่ยวข้องกับเนื้อสัตว์ที่เพาะเลี้ยง การตรวจสอบมักใช้เวลา 7–10 วัน, โดยโรงงานที่มีประสิทธิภาพสูงสามารถทำเวลาได้ต่ำกว่า 7 วัน [2]. ระบบบันทึกข้อมูลแบบอิเล็กทรอนิกส์สามารถเร่งกระบวนการตรวจสอบเหล่านี้ได้อย่างมาก เช่น ระบบที่ผสานเข้ากับ ระบบการผลิตเนื้อสัตว์เพาะเลี้ยง, - ลดเวลาลงครึ่งหนึ่งเมื่อเทียบกับวิธีการใช้กระดาษ - ตราบใดที่ได้รับการตรวจสอบเพื่อให้เป็นไปตามข้อกำหนดของ Part 11 และรักษาความสมบูรณ์ของข้อมูล [1].
สิ่งที่บริษัทเนื้อสัตว์เพาะเลี้ยงที่ได้รับการอนุมัติจาก FDA ทำได้ดี
บริษัทเนื้อสัตว์เพาะเลี้ยงที่ได้รับการอนุมัติจาก FDA ได้ตั้งมาตรฐานสูงโดยการนำแนวปฏิบัติที่แก้ไขปัญหาด้านเอกสารและปฏิบัติตามมาตรฐานความปลอดภัยที่เข้มงวด
เมื่อ UPSIDE Foods กลายเป็นบริษัทเนื้อสัตว์เพาะเลี้ยงแห่งแรกที่ผ่านการปรึกษาก่อนการตลาดของ FDA ในเดือนพฤศจิกายน 2022 พวกเขาได้สร้างแบบอย่างให้กับอุตสาหกรรมองค์การอาหารและยา (FDA) ได้ออกจดหมาย "ไม่มีคำถามเพิ่มเติม" หลังจากการตรวจสอบกระบวนการผลิตอย่างละเอียด ซึ่งรวมถึงการจัดตั้งสายเซลล์ ธนาคารเซลล์ การควบคุมการผลิต และส่วนประกอบและอินพุตทั้งหมด [16]. ความสำเร็จนี้เน้นย้ำถึงความสำคัญของการจัดทำเอกสารอย่างละเอียดในการปฏิบัติตามข้อกำหนดที่เข้มงวดของ FDA
การปฏิบัติตามมาตรฐานความปลอดเชื้อและการปฏิบัติตามข้อกำหนด
ความสำเร็จที่โดดเด่นของ UPSIDE Foods คือวิธีการที่ละเอียดถี่ถ้วนในการติดตามแหล่งที่มาของอินพุต ส่วนประกอบการผลิตทุกชิ้นได้รับการบันทึกอย่างละเอียด เพื่อให้มั่นใจถึงความรับผิดชอบที่ชัดเจนตั้งแต่สายเซลล์เริ่มต้นจนถึงผลิตภัณฑ์สุดท้าย [16]. ระดับความโปร่งใสนี้ทำให้ผู้ตรวจสอบของ FDA สามารถติดตามทุกขั้นตอนของกระบวนการผลิต ยืนยันว่ามาตรฐานความปลอดภัยทั้งหมดได้รับการปฏิบัติตามอย่างสม่ำเสมอ
"การปรึกษาก่อนการตลาดของ FDA กับบริษัทได้รวมถึงการประเมินกระบวนการผลิตของบริษัทและวัสดุเซลล์ที่เพาะเลี้ยงที่ผลิตโดยกระบวนการผลิต รวมถึงการจัดตั้ง สายเซลล์หลักและสายเซลล์ที่ทำให้เป็นอมตะ และธนาคารเซลล์ การควบคุมการผลิต และส่วนประกอบและอินพุตทั้งหมด" – U.S. สำนักงานคณะกรรมการอาหารและยา [16]
บริษัทที่ประสบความสำเร็จอื่น ๆ ได้ทำตามโดยการนำเอกสารกระบวนการปลอดเชื้อที่มีรายละเอียดมาใช้ ซึ่งรวมถึงขั้นตอนสำคัญเช่น ขั้นตอนการสวมชุดและการจัดการแบบปลอดเชื้อ [3]. ต่างจากความล้มเหลวของเอกสารก่อนหน้านี้ บริษัทเหล่านี้ได้ใช้ระบบการตรวจสอบแบบชั้น ซึ่งรวมถึงการตรวจสอบโดยผู้ปฏิบัติงาน การควบคุมการผลิต และการตรวจสอบโดยหน่วยคุณภาพ เพื่อจับข้อผิดพลาดที่อาจเกิดขึ้นก่อนการปล่อยชุดผลิตภัณฑ์ [15]. ระบบบันทึกแบทช์อิเล็กทรอนิกส์ยังมีบทบาทสำคัญในการบังคับให้มีการลงนามที่จำเป็นในแต่ละขั้นตอนและรักษาร่องรอยการตรวจสอบที่ไม่สามารถเปลี่ยนแปลงได้ตามข้อกำหนดของ 21 CFR Part 11 [3][2].
แนวทางปฏิบัติที่เข้มงวดเหล่านี้ขยายไปสู่การจัดการกับการเบี่ยงเบนและความล้มเหลวของบริษัทต่างๆ อย่างเป็นธรรมชาติ
กระบวนการ CAPA สำหรับความล้มเหลวของแบทช์
เมื่อแบทช์ไม่เป็นไปตามข้อกำหนด บริษัทที่ได้รับการอนุมัติจาก FDA จะดำเนินการอย่างรวดเร็วและเป็นระบบ กระบวนการแก้ไขและป้องกัน (CAPA) ของพวกเขารวมถึงการวิเคราะห์สาเหตุรากฐานอย่างเป็นทางการ การประเมินผลกระทบ และการดำเนินการแก้ไขที่มีการบันทึกไว้อย่างชัดเจน [3]. การเบี่ยงเบนใดๆ จะได้รับการจัดการภายในกรอบการประกันคุณภาพแบบบูรณาการ เพื่อให้มั่นใจว่าปัญหาทั้งหมดได้รับการตรวจสอบอย่างละเอียด มีเหตุผล และบันทึกไว้ก่อนที่จะดำเนินการผลิตต่อ [2].
มองไปข้างหน้า ความสมบูรณ์ของข้อมูลจะเป็นจุดสนใจหลักของการบังคับใช้กฎหมายของ FDA สำหรับปี 2024–2025 [1].
วิธีปรับปรุงการปฏิบัติด้านบันทึกแบทช์ของคุณ
การเสริมสร้างการปฏิบัติด้านบันทึกแบทช์ต้องการการบันทึกที่แม่นยำเพื่อแก้ไขข้อบกพร่องทั่วไปที่มักพบในระหว่างการตรวจสอบของ FDA นี่คือกลยุทธ์บางประการในการจัดการกับความท้าทายหลัก
ดำเนินการตรวจสอบบันทึกแบทช์อย่างอิสระ
การตรวจสอบจากบุคคลที่สามเป็นประจำสามารถเปิดเผยปัญหาที่การตรวจสอบภายในอาจมองข้าม เริ่มต้นด้วยการมุ่งเน้นไปที่ระบบที่สำคัญ เช่น ระบบการจัดการข้อมูลห้องปฏิบัติการ (LIMS), ระบบการดำเนินการผลิต (MES), และการวางแผนทรัพยากรองค์กร (ERP) ให้ความสำคัญกับเอกสารที่มีผลกระทบต่อกฎระเบียบสูง เช่น บันทึกการทดสอบการปล่อย, ข้อมูลความคงตัว, และบันทึกการผลิตแบทช์
วิธีที่มีประสิทธิภาพวิธีหนึ่งคือการทดสอบการดึงตัวอย่าง.สุ่มเลือกชุดการผลิตล่าสุดและสร้างประวัติการผลิตและห้องปฏิบัติการใหม่ ซึ่งสามารถช่วยระบุข้อมูลที่ขาดหาย ลายเซ็นที่ไม่สมบูรณ์ หรือช่องว่างในเอกสารที่อาจนำไปสู่การอ้างอิงข้อบังคับ ตรวจสอบเส้นทางการตรวจสอบที่ระบบสร้างขึ้นกับรายการที่บันทึกด้วยมือเพื่อระบุการเปลี่ยนแปลงหรือการลบที่ไม่ได้รับอนุญาต
ตรวจสอบรายงาน Out-of-Specification (OOS) และ Out-of-Trend (OOT) ทั้งหมดจากปีที่ผ่านมา ประเมินว่าการวิเคราะห์สาเหตุรากฐานมีความละเอียดถี่ถ้วนหรือไม่ และหากการดำเนินการแก้ไขและป้องกัน (CAPAs) ได้รับการดำเนินการอย่างเพียงพอหรือไม่ ควรสังเกตว่าปัญหาเอกสารคิดเป็น 21% ของจดหมายเตือนจาก FDA ในขณะที่ข้อผิดพลาดของมนุษย์มีส่วนทำให้เกิดปัญหาบันทึกชุดการผลิต 50% ในการผลิตยา [2].
"ไม่ใช่ความซับซ้อนของกระบวนการที่ทำให้เกิดการอ้างอิง - แต่เป็นความไม่สอดคล้องกัน ความไม่สมบูรณ์ และการกำกับดูแลที่ไม่ดี" – GXP Auditing & Consulting Services [5]
จำลองการตรวจสอบตามกฎระเบียบผ่านการทบทวนจำลองเป็นระยะๆ การปฏิบัตินี้ช่วยให้ทีมงานสามารถรับรู้ถึงความไม่สอดคล้องกันและปัญหาความสมบูรณ์ของข้อมูลที่อาจเกิดขึ้นก่อนการตรวจสอบจริง ตรวจสอบให้แน่ใจว่าบันทึกทั้งหมดปฏิบัติตามหลักการ ALCOA+: สามารถระบุได้, อ่านได้, ทันเวลา, ต้นฉบับ, ถูกต้อง, สมบูรณ์, สม่ำเสมอ, ทนทาน, และพร้อมใช้งาน.
เมื่อความสมบูรณ์ของเอกสารมั่นคงแล้ว ให้มุ่งเน้นไปที่การตรวจสอบคุณภาพของวัตถุดิบทั้งหมด.
ทดสอบวัตถุดิบทั้งหมดสำหรับการปนเปื้อนทางจุลชีววิทยา
การทดสอบความปลอดเชื้อและความแรงอย่างอิสระสำหรับวัตถุดิบทั้งหมดเป็นสิ่งจำเป็น - อย่าพึ่งพาเฉพาะใบรับรองการวิเคราะห์ (CoAs) จากผู้จัดจำหน่ายเท่านั้น สิ่งนี้มีความสำคัญอย่างยิ่งสำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง เนื่องจากการปนเปื้อนสามารถทำลายทั้งชุดได้
ตัวอย่างเช่น ในเดือนกุมภาพันธ์ 2013 Central Admixture Pharmacy Services ได้รับการอ้างอิงจาก FDA เนื่องจากการควบคุมจุลชีพที่ไม่เพียงพอในระหว่างการปล่อยชุดผลิตภัณฑ์ปลอดเชื้อ บริษัทต้องแนะนำขั้นตอนการควบคุมจุลชีพโดยละเอียดในขั้นตอนการปฏิบัติงานมาตรฐาน (SOPs) [4].
จุดตรวจสอบจุลชีพในกระบวนการสามารถป้องกันการพึ่งพาการทดสอบผลิตภัณฑ์ขั้นสุดท้ายมากเกินไป รวมจุดตรวจสอบเหล่านี้ใน SOPs การปล่อยชุดผลิตภัณฑ์และรักษาการบันทึกเอกสารที่เข้มงวดในขณะเดียวกัน บันทึกผลการทดสอบและขั้นตอนการผลิตทั้งหมดในขณะที่เกิดขึ้นเพื่อหลีกเลี่ยงการบันทึกย้อนหลังหรือการบันทึกที่ล่าช้า ซึ่งอาจส่งผลให้ได้รับการอ้างอิงจาก FDA
เก็บไฟล์ซัพพลายเออร์ที่ครอบคลุม รวมถึง CoAs รายงานการตรวจสอบ ข้อตกลงคุณภาพ และประวัติของการเบี่ยงเบนใด ๆ ที่เกี่ยวข้องกับวัสดุที่เข้ามา
การเสริมสร้างแนวทางปฏิบัติในการบันทึกชุดผลิตภัณฑ์เพิ่มเติมเกี่ยวข้องกับการปรับกระบวนการให้สอดคล้องกับมาตรฐานที่กำหนด เช่น HACCP และ GCCP
จัดทำบันทึกให้สอดคล้องกับมาตรฐาน HACCP และ GCCP
การนำหลักการวิเคราะห์อันตรายและจุดควบคุมวิกฤต (HACCP) มาใช้ในบันทึกการผลิตช่วยให้มั่นใจได้ว่าตัวแปรกระบวนการที่สำคัญได้รับการตรวจสอบและบันทึกตลอดการผลิต ซึ่งรวมถึงการจัดตั้งจุดตรวจสอบการทดสอบจุลชีพในกระบวนการ แทนที่จะพึ่งพาการทดสอบในขั้นตอนสุดท้ายเพียงอย่างเดียว
สำหรับผู้ผลิตเนื้อสัตว์ที่เพาะเลี้ยง การปฏิบัติตามมาตรฐานการปฏิบัติการเพาะเลี้ยงเซลล์ที่ดี (GCCP) มีความสำคัญเท่าเทียมกัน บันทึกการผลิตควรรวมถึงรายละเอียดของการจัดการปลอดเชื้อ ขั้นตอนการสวมชุด และการตรวจสอบสิ่งแวดล้อมที่เชื่อมโยงกับเกณฑ์การปล่อยชุด[3][4]. ขั้นตอนเหล่านี้ช่วยรักษาการปฏิบัติตามข้อกำหนดและรับรองความปลอดภัยของผลิตภัณฑ์
ข้อมูลอุตสาหกรรมแสดงให้เห็นว่า 52% ของการละเมิดเอกสารเพิ่มขึ้นเมื่อไม่มีซอฟต์แวร์การผลิตชุดที่เหมาะสม[3][4]. กรณีตัวอย่าง: ในเดือนกุมภาพันธ์ 2023 ศูนย์ผสมยาปลอดเชื้อ Nephron ได้รับการสังเกตจาก FDA เนื่องจากขาดขั้นตอนการควบคุมในการตรวจสอบตัวแปรกระบวนการที่สำคัญก่อนการปล่อยชุด [4]. สิ่งนี้เน้นย้ำถึงความจำเป็นในการจัดทำเอกสารเชิงรุกที่สอดคล้องกับมาตรฐานที่ได้รับการยอมรับ
การเปลี่ยนไปใช้บันทึกชุดอิเล็กทรอนิกส์ (EBR) สามารถลดข้อผิดพลาดในการจัดทำเอกสารได้อย่างมาก - มากถึง 50% - ผ่านการเก็บข้อมูลแบบเรียลไทม์และเวิร์กโฟลว์อัตโนมัติ [2]. ระบบเหล่านี้จะระบุผลการทดสอบจุลชีพที่ขาดหายไปหรือการตรวจสอบที่ไม่สมบูรณ์ก่อนที่ชุดจะก้าวหน้า ลดข้อผิดพลาดของมนุษย์
"FDA คาดหวังให้บันทึกเป็น ALCOA(+): สามารถระบุได้, อ่านได้, ทันเวลา, ต้นฉบับ, ถูกต้อง - รวมถึง สมบูรณ์, สม่ำเสมอ, ทนทาน, และพร้อมใช้งาน" – Atlas Compliance [1]
ความคลาดเคลื่อนหรือการเบี่ยงเบนที่ไม่สามารถอธิบายในบันทึกชุดการผลิตควรเชื่อมโยงกับการสอบสวนอย่างเป็นทางการและระบบ CAPA จำกัดสิทธิ์ในการเขียนและลบเพื่อปกป้องความสมบูรณ์ของข้อมูลการทดสอบจุลชีววิทยาอิเล็กทรอนิกส์ ผู้ผลิตที่มีการแข่งขันมุ่งหมายที่จะตรวจสอบและปล่อย 95% ของชุดการผลิตภายใน 30 วันหลังจากการผลิตเสร็จสิ้น [2].
การดำเนินการเหล่านี้ไม่เพียงแต่ลดความเสี่ยงของการถูกอ้างอิง แต่ยังสอดคล้องกับมาตรฐานการบันทึกเอกสารที่เข้มงวดซึ่งเน้นในระหว่างการตรวจสอบของ FDA ล่าสุด
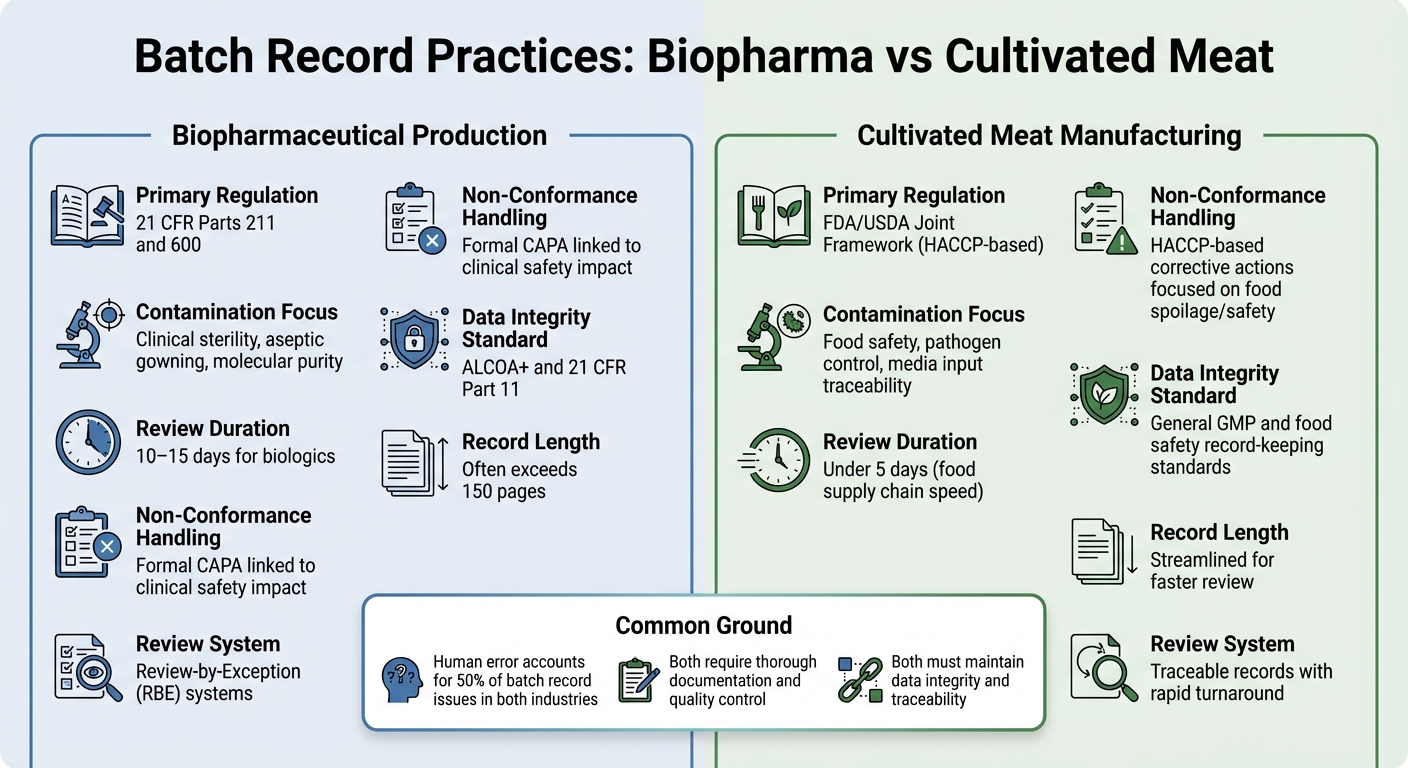
ชีวเภสัชภัณฑ์ vs เนื้อสัตว์เพาะเลี้ยง: ความแตกต่างของบันทึกการผลิต
การเปรียบเทียบข้อกำหนดบันทึกการผลิตระหว่างชีวเภสัชภัณฑ์และเนื้อสัตว์เพาะเลี้ยง
การพิจารณาความแตกต่างในแนวทางปฏิบัติของบันทึกการผลิตระหว่างการผลิตชีวเภสัชภัณฑ์และการผลิตเนื้อสัตว์เพาะเลี้ยง ช่วยให้เห็นภาพที่ชัดเจนขึ้นว่าข้อกำหนดด้านกฎระเบียบมีผลต่อการจัดลำดับความสำคัญของเอกสารในอุตสาหกรรมเหล่านี้อย่างไร
ทั้งสองภาคส่วนต้องการเอกสารที่ละเอียดถี่ถ้วน แต่กรอบการกำกับดูแลและวัตถุประสงค์ในการควบคุมของพวกเขาแตกต่างกันอย่างมาก ในชีวเภสัชภัณฑ์ บันทึกการผลิตถูกควบคุมอย่างเข้มงวดภายใต้21 CFR Parts 211 and 600, ซึ่งกำหนดให้หน่วยควบคุมคุณภาพต้องตรวจสอบและอนุมัติบันทึกการผลิตและการควบคุมทั้งหมดก่อนที่จะสามารถปล่อยชุดผลิตภัณฑ์ได้ [2]. ในทางกลับกัน ผู้ผลิตเนื้อสัตว์เพาะเลี้ยงมักจะปฏิบัติตามมาตรฐานHACCP และ GCCP ข้อความเหล่านี้เน้นไปที่ความปลอดภัยของอาหารและการควบคุมเชื้อโรค มากกว่าความปลอดเชื้อในระดับคลินิกที่ต้องการสำหรับชีววิทยาฉีดเข้าเส้นเลือด
บันทึกการผลิตของไบโอฟาร์มามักจะมีความยาว บางครั้งเกินกว่า 150 หน้า และกระบวนการตรวจสอบอาจใช้เวลา 10–15 วัน เพื่อให้กระบวนการนี้มีประสิทธิภาพมากขึ้น บริษัทไบโอฟาร์มาหลายแห่งใช้ระบบ Review-by-Exception (RBE) ซึ่งสรุปความเบี่ยงเบนที่สำคัญลงในรายงานที่กระชับ ในขณะเดียวกัน ผู้ผลิตเนื้อสัตว์เพาะเลี้ยงมุ่งเน้นไปที่บันทึกที่สามารถตรวจสอบได้ภายในเวลาน้อยกว่าห้าวัน สะท้อนถึงความเร็วของห่วงโซ่อุปทานอาหาร [2].
เนื้อหาของบันทึกเหล่านี้ยังเน้นถึงความสำคัญที่แตกต่างกัน การตรวจสอบไบโอฟาร์มามักจะเน้นไปที่รายละเอียดการประมวลผลปลอดเชื้อ เช่น ขั้นตอนการสวมใส่เสื้อผ้าและการควบคุมสิ่งแวดล้อม ในทางตรงกันข้าม บันทึกของเนื้อสัตว์เพาะเลี้ยงต้องเน้นที่การป้อนสื่อและการทดสอบทางจุลชีววิทยาเพื่อให้มั่นใจในความปลอดภัยของอาหารสำหรับเนื้อสัตว์ที่เพาะเลี้ยง ความท้าทายอยู่ที่การติดตามสื่อที่ซับซ้อน การบันทึกการทดสอบทางจุลชีววิทยาสำหรับวัสดุทั้งหมด และการปฏิบัติตามขีดจำกัดความปลอดภัยของอาหารที่สำคัญ - โดยไม่ต้องปฏิบัติตามข้อกำหนดความปลอดเชื้อที่เข้มงวดของยา
แนวโน้มการปนเปื้อนและการไม่เป็นไปตามข้อกำหนด
| คุณสมบัติ | การผลิตชีวเภสัชภัณฑ์ | การผลิตเนื้อสัตว์เพาะเลี้ยง |
|---|---|---|
| กฎระเบียบหลัก | 21 CFR Parts 211 and 600 [2] | กรอบการทำงานร่วมของ FDA/USDA (ตาม HACCP) |
| การเน้นการปนเปื้อน | ความปลอดเชื้อทางคลินิก, การสวมชุดปลอดเชื้อ, ความบริสุทธิ์ระดับโมเลกุล[2] | ความปลอดภัยของอาหาร, การควบคุมเชื้อโรค, การตรวจสอบย้อนกลับของสื่อที่ใช้ |
| ระยะเวลาการตรวจสอบ | 10–15 วันสำหรับชีววัตถุ[2] | ต่ำกว่า 5 วัน (ความเร็วของห่วงโซ่อุปทานอาหาร) |
| การจัดการการไม่เป็นไปตามข้อกำหนด | การดำเนินการแก้ไข CAPA อย่างเป็นทางการที่เชื่อมโยงกับผลกระทบด้านความปลอดภัยทางคลินิก [2] | การดำเนินการแก้ไขตาม HACCP ที่มุ่งเน้นการเน่าเสีย/ความปลอดภัยของอาหาร |
| มาตรฐานความสมบูรณ์ของข้อมูล | ALCOA+ และ 21 CFR Part 11 [1] | มาตรฐานการเก็บบันทึก GMP ทั่วไปและความปลอดภัยของอาหาร |
ในขณะที่อัตราความผิดพลาดของมนุษย์มีความคล้ายคลึงกันในทั้งสองอุตสาหกรรม - ประมาณ 50% ของปัญหาบันทึกชุดเกิดจากความผิดพลาดของมนุษย์ [2] - แต่ความเสี่ยงนั้นแตกต่างกันในอุตสาหกรรมชีวเภสัชภัณฑ์ แม้แต่การเบี่ยงเบนที่ไม่ได้บันทึกเพียงครั้งเดียวก็อาจมีผลกระทบอย่างร้ายแรงต่อความปลอดภัยของผู้ป่วย สำหรับเนื้อสัตว์ที่เพาะเลี้ยง ความเสี่ยงของการปนเปื้อนเกี่ยวข้องกับเชื้อโรคที่เกิดจากอาหารและการเน่าเสีย ซึ่งอาจส่งผลต่อการผลิตทั้งหมด
บทสรุป
บันทึกการผลิตทำหน้าที่เป็นบันทึกอย่างเป็นทางการสำหรับการผลิตเนื้อสัตว์ที่เพาะเลี้ยงทุกครั้ง - หากขั้นตอนไม่ได้ถูกบันทึกไว้ หน่วยงานกำกับดูแลจะถือว่าไม่ได้ดำเนินการ [6] [3]. สิ่งนี้เน้นย้ำถึงความสำคัญของการบันทึกที่แม่นยำและการควบคุมคุณภาพอย่างเข้มงวด
การตรวจสอบของ FDA เน้นย้ำว่าความสมบูรณ์ของข้อมูลต้องสอดคล้องกับหลักการ ALCOA+ [1]. ทีมควบคุมคุณภาพจำเป็นต้องตรวจสอบและอนุมัติบันทึกการผลิตทั้งหมดก่อนที่จะปล่อยชุดผลิตภัณฑ์ [2][17], และการเบี่ยงเบนใด ๆ ต้องได้รับการตรวจสอบอย่างรวดเร็วพร้อมการวิเคราะห์สาเหตุที่แท้จริงที่มีการบันทึกไว้ [2][5]. แม้ว่าความผิดพลาดของมนุษย์จะคิดเป็น 50% ของปัญหาบันทึกชุดผลิตภัณฑ์ แต่การตรวจสอบสองระดับและกระบวนการ CAPA (การดำเนินการแก้ไขและป้องกัน) ที่มีโครงสร้างสามารถช่วยลดความเสี่ยงเหล่านี้ได้ [2][5].
"ไม่ใช่ความซับซ้อนของกระบวนการที่ทำให้เกิดการอ้างอิง - แต่เป็นความไม่สม่ำเสมอ ความไม่สมบูรณ์ และการควบคุมที่ไม่ดี" - GXP Auditing & Consulting Services [5]
เพื่อเอาชนะความท้าทายเหล่านี้ ผู้ผลิตเนื้อสัตว์เพาะเลี้ยงควรมุ่งเน้นไปที่การตรวจสอบอิสระ การทดสอบอย่างเข้มงวดของ ส่วนผสมที่ปลอดภัยต่ออาหาร สำหรับการปนเปื้อนทางจุลชีววิทยา และการรับรองเอกสารให้เป็นไปตามมาตรฐาน HACCP และ GCCP การนำระบบบันทึกแบทช์อิเล็กทรอนิกส์ที่ผ่านการตรวจสอบภายใต้ 21 CFR Part 11 [1], สามารถลดข้อผิดพลาดและเร่งกระบวนการตรวจสอบได้อย่างมาก
สภาพแวดล้อมด้านกฎระเบียบต้องการความแม่นยำ แต่สามารถนำทางได้โดยการเรียนรู้จากข้อผิดพลาดของอุตสาหกรรมชีวเภสัช เช่น การขาดลายเซ็นที่ Qinhuangdao Zizhu Pharmaceutical [17], การตรวจสอบสองชั้นที่ไม่เพียงพอที่ Terumo Corp [18] , และเอกสารการเบี่ยงเบนที่ไม่เพียงพอที่ Torrent Pharmaceuticals [18] - บริษัทเนื้อสัตว์เพาะเลี้ยงสามารถสร้างระบบที่สอดคล้องตั้งแต่เริ่มต้น การนำบทเรียนเหล่านี้มาใช้จะช่วยให้เกิดการปฏิบัติตามข้อกำหนดอย่างรอบคอบและคุณภาพที่สม่ำเสมอ การเก็บรักษาบันทึกอย่างปลอดภัย การรายงานการเบี่ยงเบนอย่างทันท่วงที และการดำเนินการตรวจสอบจำลองที่สมจริงจะช่วยให้บันทึกการผลิตพร้อมสำหรับการตรวจสอบและการผลิตสามารถตรวจสอบย้อนกลับได้อย่างเต็มที่
สำหรับทรัพยากรเพิ่มเติมและคำแนะนำจากผู้เชี่ยวชาญเกี่ยวกับการรักษามาตรฐานการผลิตสูงในอุตสาหกรรมการผลิตเนื้อสัตว์เพาะเลี้ยง โปรดเยี่ยมชม
คำถามที่พบบ่อย
บันทึกการผลิตสำหรับเนื้อสัตว์ที่เพาะเลี้ยงควรมีอะไรบ้าง?
บันทึกการผลิตสำหรับเนื้อสัตว์ที่เพาะเลี้ยงทำหน้าที่เป็นบันทึกที่ครอบคลุมของกระบวนการผลิตทั้งหมด ซึ่งต้องรวมถึง คำแนะนำในการประมวลผลอย่างละเอียด, บันทึกการดำเนินการทีละขั้นตอน, และบันทึก การเบี่ยงเบน ที่เกิดขึ้นระหว่างการผลิต นอกจากนี้ ควรบันทึก การทดสอบระหว่างกระบวนการ และ การทดสอบการปล่อยผลิตภัณฑ์ เพื่อยืนยันว่าผลิตภัณฑ์ตรงตามมาตรฐานความปลอดภัย คุณภาพ และข้อกำหนดทางกฎหมาย
เราจะพิสูจน์ความปลอดเชื้อโดยใช้บันทึกการผลิตได้อย่างไร?
การพิสูจน์ความปลอดเชื้อผ่านบันทึกการผลิตเกี่ยวข้องกับการตรวจสอบขั้นตอนการฆ่าเชื้อที่บันทึกไว้ ผลการทดสอบ และรายงานการควบคุมคุณภาพของสื่ออย่างละเอียดเพื่อให้แน่ใจว่าตรงตามข้อกำหนดทางกฎหมายเป็นสิ่งสำคัญที่จะต้องจัดการกับความเบี่ยงเบนหรือการทดสอบที่ล้มเหลวผ่านการสืบสวนอย่างละเอียดและ CAPAs (การดำเนินการแก้ไขและป้องกัน) กระบวนการนี้ทำให้มั่นใจได้ว่าทุกขั้นตอนถูกปฏิบัติตามและปัญหาต่างๆ ได้รับการแก้ไขอย่างถูกต้องเพื่อรักษามาตรฐานความปลอดเชื้อ
เมื่อใดที่จำเป็นต้องมีบันทึกชุดงานอิเล็กทรอนิกส์ (Part 11)?
บันทึกชุดงานอิเล็กทรอนิกส์มีความจำเป็นภายใต้ Part 11 เมื่อมีการใช้ระบบอิเล็กทรอนิกส์ในการบันทึก สืบสวน และให้เหตุผลเกี่ยวกับความเบี่ยงเบนของบันทึกชุดงาน พวกเขามีบทบาทสำคัญในการรับรองการปฏิบัติตาม 21 CFR Part 211.192, การปกป้องความสมบูรณ์ของข้อมูล การปฏิบัติตามกำหนดเวลาการสืบสวน และการรับรองการกำกับดูแลการจัดการที่มีประสิทธิภาพ









