

หากคุณแก้ไขก่อนและตรวจสอบภายหลัง คุณสามารถแก้ไขการเปลี่ยนแปลงที่ไม่ตรงเป้าหมายในโคลนได้ ฉันจะรักษากระบวนการทำงานให้เรียบง่าย: เลือกวิธีการแก้ไขที่มีความเสี่ยงต่ำที่สุด, ลดการเปิดรับของบรรณาธิการให้สั้น, และทดสอบทั้งไซต์ที่คาดว่าจะไม่ตรงเป้าหมายและความเสถียรของโคลนก่อนการปล่อย
สำหรับวิศวกรกระบวนการชีวภาพ, นักวิทยาศาสตร์เพาะเลี้ยงเซลล์, และทีมวิจัยและพัฒนาของเนื้อสัตว์ที่เพาะเลี้ยง R&D ประเด็นหลักคือชัดเจน ระบบ CRISPR ยังสามารถตัดที่ไซต์ที่ใกล้เคียงกันได้, มักจะมี 3–6 ความไม่ตรงกัน ที่ยอมรับได้, และข้อผิดพลาดเหล่านั้นสามารถส่งต่อไปยังโคลนเซลล์เดี่ยวที่ขยายตัวได้ บทความนี้แบ่งการควบคุมความเสี่ยงออกเป็นสามขั้นตอน: ก่อนการแก้ไข, ระหว่างการแก้ไข, และ หลังการแก้ไข.
นี่คือรายการตรวจสอบทั้งหมดในรูปแบบที่เข้าใจง่าย:
-
เลือกเครื่องมือแก้ไขที่มีความเสี่ยงต่ำที่สุดสำหรับงาน
- ใช้ การแก้ไขฐาน หรือ การแก้ไขหลัก เมื่อสามารถทำการแก้ไขได้โดยไม่ต้องตัดสายคู่
- ใช้ การปรับเปลี่ยนที่ใช้ dCas9 หากคุณต้องการเพียงการควบคุมยีน
- หากคุณต้องการนิวคลีเอส ให้เริ่มด้วย Cas9 ที่มีความแม่นยำสูง รุ่น
-
ล็อควัสดุเริ่มต้น
- ยืนยัน ตัวตนของสายเซลล์
- ตรวจสอบ ไมโคพลาสมา
- บันทึก หมายเลขการผ่าน
- จัดลำดับ ตำแหน่งเป้าหมายจริง ในสายการทำงาน ไม่ใช่แค่จีโนมอ้างอิง
-
คัดกรองไกด์ก่อนทำงานเปียก
- ใช้ เครื่องมือที่ใช้การจัดแนว และ เครื่องมือที่ใช้การให้คะแนน สำหรับการตรวจสอบเป้าหมายที่ไม่ต้องการร่วมกัน
- เลือกไกด์ที่มีโปรไฟล์เป้าหมายที่ไม่ต้องการที่สะอาดกว่าแทนที่จะเลือกที่มีแค่กิจกรรมเป้าหมายที่สูงกว่า
- ระวังความยาวของไกด์, เนื้อหา GC 40–60%, และการวิ่งของโฮโมโพลีเมอร์
-
จำกัดการสัมผัสภายในเซลล์
- ใช้ RNP หรือ mRNA แทนการใช้ระบบพลาสมิดหรือไวรัสถ้าเป็นไปได้
- ใช้ ขนาดยาที่มีประสิทธิภาพต่ำสุด
- หลีกเลี่ยงการขยายเวลาการคงอยู่ของตัวแก้ไขเพียงเพื่อบังคับผลการถ่ายโอน
-
เพิ่มการควบคุมเพิ่มเติมสำหรับกรณีที่มีความเสี่ยงสูง
- พิจารณา paired nickases
- ใช้ ระบบที่สามารถกระตุ้นได้, split-Cas9, หรือ ระบบที่ควบคุมด้วยแสง เมื่อเวลามีความสำคัญ
- เพิ่ม โปรตีนต้าน CRISPR เป็นขั้นตอนการปิดเมื่อจำเป็น
-
ตรวจสอบให้ถูกต้องหลังจากแก้ไข
- ยืนยัน การแก้ไขที่ตรงเป้าหมาย ก่อน
- ตรวจสอบทุกตำแหน่งที่คาดว่าจะไม่ตรงเป้าหมายด้วย การทำ NGS ของแอมพลิคอนที่กำหนดเป้าหมาย
- ย้ายไปที่ GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, หรือ WGS เมื่อความเสี่ยงของโครงการสูงขึ้น
- สำหรับ base หรือ prime editors , เพิ่ม การตรวจสอบระดับ RNA ที่เกี่ยวข้อง
-
อย่าปล่อยโคลนเดียวโดยอิงจากลำดับเพียงอย่างเดียว
- เปรียบเทียบ โคลนที่เป็นอิสระ 2–3 โคลน
- ใช้ การควบคุมพ่อแม่ที่ไม่ได้แก้ไข
- ลบโคลนที่มีความไม่เสถียรหรือการเปลี่ยนแปลงฟีโนไทป์
- ปล่อยเฉพาะเมื่อสถานะแก้ไข, การตรวจสอบนอกเป้าหมาย, และบันทึกทั้งหมดเสร็จสมบูรณ์
วิธีคิดแบบสั้น ๆ: ออกแบบเพื่อหลีกเลี่ยงการตัดที่ไม่ตรงเป้าหมาย ส่งมอบเพื่อจำกัดเวลาในเซลล์ จากนั้นตรวจสอบความถูกต้องในระดับที่ความเสี่ยงของโครงการกำหนด. นั่นคือหัวข้อที่เชื่อมโยงทั้งชิ้นงานเข้าด้วยกัน
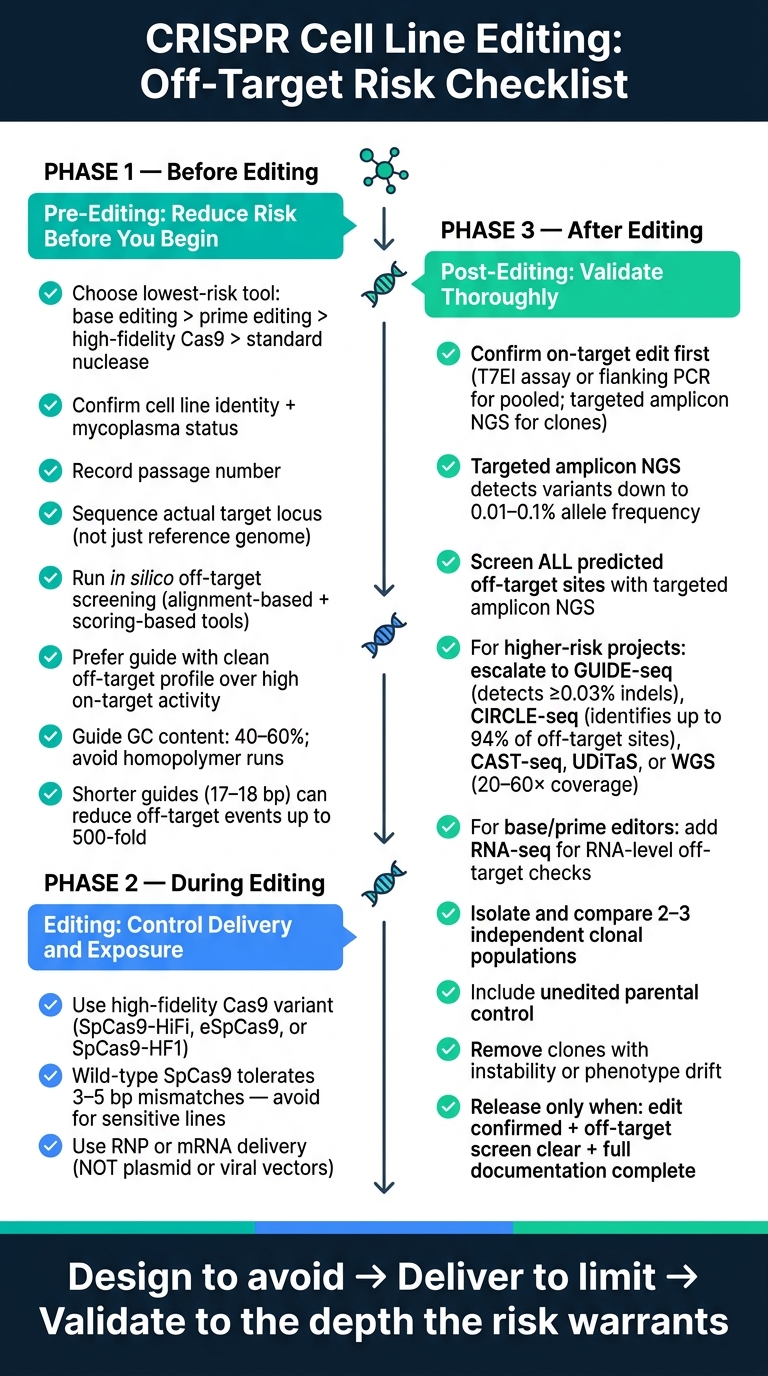
การควบคุมความเสี่ยงนอกเป้าหมายของ CRISPR: รายการตรวจสอบ 3 ขั้นตอนสำหรับการแก้ไขสายเซลล์
รายการตรวจสอบก่อนการแก้ไข: ลดความเสี่ยงก่อนเริ่มการแก้ไข
กำหนดเป้าหมายการแก้ไขและเลือกวิธีการแก้ไขที่มีความเสี่ยงต่ำที่สุด
ก่อนที่คุณจะสั่งซื้อสารเคมีใด ๆ ให้ชัดเจนมากว่าการแก้ไขนั้นมีจุดประสงค์เพื่อทำอะไร การลบออก การเพิ่มเข้า การเปลี่ยนแปลงนิวคลีโอไทด์เดี่ยว และการปรับเปลี่ยนการถอดรหัส ไม่ มีความเสี่ยงนอกเป้าหมายเท่ากัน พวกเขายังไม่ต้องการเครื่องมือเดียวกัน
ลำดับความเสี่ยงโดยรวมเป็นเรื่องง่าย นิวเคลียสที่สร้าง DSB เช่น Cas9 และ Cas12 อยู่ที่ปลายสุดของความเสี่ยงเพราะพวกเขาสามารถทำให้เกิดการลบขนาดใหญ่ การย้ายตำแหน่ง และการตอบสนองต่อความเสียหายของ DNA [1] [7]. ตัวแก้ไขฐานและตัวแก้ไขหลักใช้ nickases ดังนั้นพวกเขาหลีกเลี่ยง DSBs และลดความเสี่ยงของการเปลี่ยนแปลงโครงสร้าง [1][5]. สำหรับการปรับเปลี่ยนการถอดรหัส ตัวแก้ไข epigenetic เช่น dCas9 ที่ผสานกับตัวปรับเปลี่ยนการถอดรหัสจะไม่เปลี่ยนแปลงลำดับ DNA [1].
กฎปฏิบัตินั้นง่าย: ใช้วิธีที่ มีความเป็นพิษต่อยีนต่ำที่สุด ที่ยังสามารถส่งมอบการแก้ไขที่คุณต้องการได้ สำหรับการเปลี่ยนแปลงนิวคลีโอไทด์เดี่ยว CBEs หรือ ABEs เหมาะสมกว่าการใช้ HDR ซึ่งยังสามารถแนะนำ indels ได้ [3] [1]. สำหรับการแทนที่และการแทรกหรือการลบขนาดเล็ก การแก้ไขหลักมักแสดงกิจกรรมที่ไม่ตรงเป้าหมายต่ำกว่ามาตรฐาน CRISPR-Cas9 [1]. หากคุณต้องใช้เอนไซม์นิวคลีเอส ให้เลือกใช้ชนิดที่มีความแม่นยำสูง เช่น SpCas9-HiFi, eSpCas9, หรือ SpCas9-HF1 [1] [6].
เมื่อกำหนดวิธีการแก้ไขแล้ว ให้ล็อคสายเซลล์ที่ทำงานและลำดับเป้าหมายที่แน่นอน
ยืนยันตัวตนของสายเซลล์ ประวัติ และลำดับตำแหน่งเป้าหมาย
หากสายเซลล์ถูกระบุผิดหรือมีการปนเปื้อนข้าม ส่วนที่เหลือของกระบวนการทำงานจะเริ่มสั่นคลอน แม้แต่ RNA ไกด์ที่ออกแบบมาอย่างดีก็ไม่สามารถกู้คืนวัสดุเริ่มต้นที่ไม่ดีได้ ตรวจสอบตัวตนของสายเซลล์ก่อนเริ่มการแก้ไข ในขณะเดียวกัน ยืนยันสถานะไมโคพลาสมาและบันทึกหมายเลขการผ่านปัจจุบัน เนื่องจากเซลล์ที่ผ่านการผ่านสูงสามารถเปลี่ยนความเสถียรของจีโนมและประสิทธิภาพการแก้ไข [1][6].
ที่สำคัญพอๆ กัน อย่าพึ่งพาเฉพาะจีโนมอ้างอิงเท่านั้น เรียงลำดับ ตำแหน่งเป้าหมายที่แน่นอน ในสายเซลล์ที่ทำงาน ขั้นตอนนี้ช่วยให้คุณระบุ SNPs หรือ indels ที่อาจขัดขวางการจับของไกด์หรือสร้างไซต์นอกเป้าหมายใหม่ [1] [6].
หลังจากนั้น ให้เข้าสู่การออกแบบไกด์
ทำการคัดกรองนอกเป้าหมายในซิลิโกก่อนเลือกสารเคมี
เมื่อยืนยันตำแหน่งเป้าหมายแล้ว ให้คัดกรอง RNA ไกด์ผู้สมัครในซิลิโกก่อนที่คุณจะเริ่มงานในห้องปฏิบัติการ ใช้ทั้งเครื่องมือที่ใช้การจัดตำแหน่ง เช่น Cas-OFFinder หรือ FlashFry , และเครื่องมือที่ใช้การให้คะแนน เช่น การให้คะแนน CFD หรือ DeepCRISPR. กลุ่มแรกช่วยค้นหาไซต์จีโนมที่มีความคล้ายคลึงกันของลำดับ กลุ่มที่สองช่วยจัดอันดับไซต์เหล่านั้นตามความน่าจะเป็นในการตัดที่คาดการณ์ไว้ [1][5].
เมื่อมีการคัดเลือกไกด์ โปรไฟล์ที่สะอาดจากการนอกเป้าหมายควรจะดีกว่าประสิทธิภาพในเป้าหมายที่ดิบ ไกด์ที่มีประสิทธิภาพในเป้าหมาย 70% และไม่มีการคาดการณ์นอกเป้าหมายเป็นจุดเริ่มต้นที่ปลอดภัยกว่าที่มีประสิทธิภาพ 90% และมีหลายไซต์ที่มีความเสี่ยงสูง [6]. ในบางกรณี การลดความยาวของไกด์จาก 20 bp เป็น 17-18 bp สามารถลดเหตุการณ์นอกเป้าหมายได้ถึง 500 เท่าโดยไม่สูญเสียความแม่นยำในเป้าหมายมากนัก [5]. ตั้งเป้าหมายให้มีเนื้อหา GC ระหว่าง 40% ถึง 60% และหลีกเลี่ยงการมีฐานที่เหมือนกันสี่ตัวหรือมากกว่าติดต่อกัน [6][5].
อย่างไรก็ตาม การคัดกรองในซิลิโกมีขีดจำกัด มันไม่สามารถคำนึงถึงสถานะโครมาติน วัฏจักรเซลล์ หรือบริบทเฉพาะของเซลล์ได้ดี [1][6][4]. คิดว่าเป็นตัวกรอง ไม่ใช่หลักฐานมันจำกัดขอบเขต แต่ไม่ได้แทนที่การยืนยันเชิงทดลอง
นำไซต์ที่มีความเสี่ยงสูงที่สุดที่คาดการณ์ไว้เข้าสู่แผนการแก้ไขและการตรวจสอบ
sbb-itb-ffee270
รายการตรวจสอบการแก้ไข: การเลือกตัวแก้ไข การส่งมอบ และการเปิดเผย
ใช้ตัวแก้ไขที่มีความจำเพาะสูงและไกด์ RNA ที่ได้รับการจัดอันดับดี
เริ่มต้นด้วยรายการสั้น ๆ ของการคาดการณ์นอกเป้าหมายและใช้มันในการเลือกตัวแก้ไข ในกรณีส่วนใหญ่ ตัวแปร SpCas9 ที่มีความแม่นยำสูง - SpCas9-HiFi, eSpCas9, หรือ SpCas9-HF1 - เป็นตัวเลือกเริ่มต้นที่ดีกว่า SpCas9 ชนิดป่า [6] [1]. SpCas9 ชนิดป่าสามารถทนต่อ การไม่ตรงกันของคู่เบสสามถึงห้า, โดยเฉพาะใน บริเวณที่ห่างจาก PAM, และนั่นสร้างความเสี่ยงนอกเป้าหมายที่มีความหมายในสายเซลล์ที่ไวต่อการสัมผัส [3].
กฎง่ายๆ ช่วยได้ที่นี่: ใช้ บรรณาธิการที่มีความแม่นยำสูงที่มีการใช้งานน้อยที่สุด ที่ยังคงให้ผลลัพธ์ตามที่ต้องการ
สำหรับบรรณาธิการพื้นฐาน ให้ติดตาม การแก้ไขที่ไม่ตั้งใจ และ ผลกระทบที่ไม่ตรงเป้าหมายของ RNA แยกจาก ความเสี่ยงที่ไม่ตรงเป้าหมายของ DNA [1] [8]. เหล่านี้เป็นโหมดความล้มเหลวที่แตกต่างกัน และต้องการการตรวจสอบแยกกัน หากคุณสามารถทำการแก้ไข โดยไม่ต้องมีการแตกหักของสายคู่, การแก้ไขฐานหรือการแก้ไขหลักอาจเหมาะสมกว่าในกระบวนการที่มีความเสี่ยงสูง [1][8].
เมื่อเลือกบรรณาธิการแล้ว งานต่อไปคือการทำให้เวลาที่อยู่ในเซลล์สั้นที่สุดเท่าที่จะเป็นไปได้
จำกัดการคงอยู่ของบรรณาธิการด้วยการส่งชั่วคราวและขนาดยาที่มีประสิทธิภาพต่ำสุด
การคงอยู่ของบรรณาธิการมีความสำคัญพอๆ กับการเลือกบรรณาธิการยิ่งตัวแก้ไขอยู่ในเซลล์นานเท่าไหร่ ก็ยิ่งมีเวลามากขึ้นในการทำงานที่ไซต์ที่มีความน่าจะเป็นต่ำ นั่นทำให้รูปแบบการส่งเป็นจุดควบคุมหลัก
ใช้ การส่งชั่วคราว เช่น RNPs หรือ mRNA, และหลีกเลี่ยง พลาสมิด DNA หรือ เวกเตอร์ไวรัส ที่ขยายการแสดงออกของตัวแก้ไข [1] [5]. ในทางปฏิบัติ การส่ง RNP ควรเป็นค่าเริ่มต้น [6] .
ปริมาณก็สำคัญเช่นกัน ความเข้มข้นของนิวคลีเอสสูงเพิ่มโอกาสในการตัดที่ไซต์นอกเป้าหมายที่มีความไวต่ำ [5]. ใช้ ปริมาณที่มีประสิทธิภาพต่ำสุด. หากประสิทธิภาพการถ่ายโอนยีนไม่ดี อย่าเพียงแค่เพิ่มสารรีเอเจนต์และหวังว่าจะดีที่สุด เพราะมักจะเปลี่ยนปัญหาแทนที่จะแก้ไข
เพิ่มมาตรการป้องกันความแม่นยำสำหรับกระบวนการที่มีความเสี่ยงสูงกว่า
กระบวนการบางอย่างต้องการมาตรการป้องกันเพิ่มเติม โดยเฉพาะอย่างยิ่งสำหรับเป้าหมายที่อยู่ใกล้ ยีนก่อมะเร็ง, ตัวยับยั้งเนื้องอก, หรือใน สายเซลล์ที่ไวต่อ p53, ซึ่งเหตุการณ์นอกเป้าหมายเพียงครั้งเดียวอาจมีค่าใช้จ่ายที่สูงเกินไป [1][6][3].
มาตรการป้องกันที่มีประโยชน์รวมถึง:
- นิกเคสคู่, ซึ่งต้องการการตัดที่อยู่ใกล้กันสองครั้ง การตัดนอกเป้าหมายเพียงครั้งเดียวมักจะถูกซ่อมแซมโดยไม่มีการกลายพันธุ์ ดังนั้นความเสี่ยงนอกเป้าหมายจึงลดลงมากเมื่อเทียบกับการตั้งค่านิวคลีเอสมาตรฐาน [4][1].
- ระบบ Inducible, light-controlled, หรือ split-Cas9, ซึ่งช่วยให้การทำงานของตัวแก้ไขอยู่ในกรอบเวลาที่จำกัดเมื่อการส่งมอบมีประสิทธิภาพและการเปิดรับต้องสั้น [1].
- โปรตีน Anti-CRISPR (Acr), ซึ่งทำหน้าที่เป็นสวิตช์ปิดการทำงาน โปรตีน Acr ที่เกิดขึ้นตามธรรมชาติเหล่านี้สามารถยกเลิกการทำงานของ CRISPR-Cas complex หลังจากช่วงเวลาที่กำหนด ทำให้คุณมีเบรกโมเลกุลในการทำงานของตัวแก้ไข [1].
รายการตรวจสอบหลังการแก้ไข: ตรวจจับเหตุการณ์นอกเป้าหมายและยืนยันโคลน
ตรวจสอบไซต์นอกเป้าหมายที่คาดการณ์ไว้ด้วยการจัดลำดับเป้าหมาย
เมื่อการแก้ไขเสร็จสิ้น ให้ยืนยันการเปลี่ยนแปลงที่ตั้งใจไว้ที่ตำแหน่งเป้าหมายก่อน สำหรับการตรวจสอบครั้งแรกอย่างรวดเร็วในเซลล์รวม คุณสามารถใช้การทดสอบการตัดแยกที่ไม่ตรงกัน เช่น T7 Endonuclease I, การย่อยข้อจำกัด หรือ PCR ที่ขนาบข้างเพียงระวังในการตีความ: แต่ละวิธีมีขีดจำกัดความไว โดยเฉพาะสำหรับการแก้ไขที่หายากหรือการเปลี่ยนแปลงแบบ homozygous [9].
สำหรับการตรวจสอบในระดับโคลน, การทำ NGS ของแอมพลิคอนที่กำหนดเป้าหมาย เป็นมาตรฐาน มันให้มุมมองเชิงปริมาณของความถี่อัลลีลและสามารถตรวจจับการเปลี่ยนแปลงได้ถึง 0.01% ถึง 0.1% [3] .
ทำการลำดับทุกตำแหน่งที่คาดว่าจะมีการเปลี่ยนแปลงนอกเป้าหมายด้วยการทำ NGS ของแอมพลิคอนที่กำหนดเป้าหมาย นั่นควรเป็นขั้นตอนการตรวจสอบเริ่มต้น
เพิ่มระดับไปยังการทดสอบทั่วทั้งจีโนมหรือโครงสร้างเมื่อความเสี่ยงของโครงการสูงขึ้น
การตรวจสอบทีละไซต์ไม่เพียงพอเสมอไป หากตัวแก้ไข, ตำแหน่งเป้าหมาย, หรือสายเซลล์บ่งบอกถึงความเสี่ยงที่ซ่อนอยู่, ให้ย้ายไปยังการทดสอบที่สามารถตรวจจับเหตุการณ์ที่คุณไม่ได้คาดการณ์ล่วงหน้าได้
การทดสอบการค้นพบทั่วทั้งจีโนม เช่น GUIDE-seq และ CIRCLE-seq ไม่จำเป็นต้องมีรายการตำแหน่งนอกเป้าหมายล่วงหน้าGUIDE-seq สามารถตรวจจับตำแหน่งนอกเป้าหมายที่มีความถี่ของ indel ต่ำถึง 0.03% [2] . CIRCLE-seq สามารถระบุได้ถึง 94% ของตำแหน่งนอกเป้าหมาย ในหลอดทดลอง [3] . วิธีการเหล่านี้มีประโยชน์เมื่อบริบทของเซลล์อาจปกปิดกิจกรรมนอกเป้าหมาย.
หากคุณกังวลเกี่ยวกับการจัดเรียงใหม่ขนาดใหญ่ การอ่านแอมพลิคอนมาตรฐานอาจพลาดปัญหาหลัก การลบ การกลับด้าน และการย้ายตำแหน่งต้องการการทดสอบที่สร้างขึ้นสำหรับการเปลี่ยนแปลงโครงสร้าง เช่น CAST-seq และ UDiTaS [1] .
การหาลำดับจีโนมทั้งหมด (WGS) เป็นตัวเลือกที่กว้างที่สุด สามารถตรวจจับ indels การเปลี่ยนแปลงโครงสร้าง และการเปลี่ยนแปลงจำนวนสำเนาทั่วทั้งจีโนม [1]. การแลกเปลี่ยนคือความลึกและค่าใช้จ่าย: โดยปกติจะต้องการความครอบคลุม 20–60× ซึ่งทำให้ไม่เหมาะสำหรับการคัดกรองประชากรจำนวนมากเป็นประจำ [1].
ใช้ targeted amplicon NGS สำหรับไซต์ที่คาดการณ์ไว้ ย้ายไปยังการทดสอบทั่วทั้งจีโนมหรือโครงสร้างสำหรับโครงการที่มีความเสี่ยงสูงกว่า สำหรับ base หรือ prime editors ให้เพิ่ม RNA-seq เพื่อตรวจสอบผลกระทบที่ไม่ตรงเป้าหมายในระดับ RNA
เลือกโคลนที่เป็นอิสระหลายตัวและบันทึกเกณฑ์การปล่อย
หลังจากการตรวจสอบลำดับ ให้ทดสอบฟีโนไทป์ในโคลนมากกว่าหนึ่งตัว
อย่าดำเนินการต่อด้วยโคลนที่แก้ไขเพียงตัวเดียว แยกและขยายประชากรโคลนที่เป็นอิสระอย่างน้อย สองถึงสาม และเปรียบเทียบกับการควบคุมพ่อแม่ที่ไม่ได้แก้ไข [4] [9]. ลบโคลนที่แสดงความไม่เสถียรหรือการเปลี่ยนแปลงฟีโนไทป์ [3]. จากนั้นยืนยันการแก้ไขที่ตั้งใจไว้ในสถานะอัลลีลที่ต้องการ ไม่ว่าจะเป็นเฮเทอโรไซกัสหรือโฮโมไซกัส โดยใช้ targeted amplicon NGS [9].
การจัดทำเอกสารไม่ใช่งานของผู้ดูแลระบบในตอนท้าย มันเป็นส่วนหนึ่งของการปล่อยโคลน บันทึก พื้นหลังของสายพันธุ์พ่อแม่, การออกแบบ sgRNA, ตัวแปรนิวเคลียส, วิธีการส่งมอบ, และผลลัพธ์ QC ทั้งหมด [3]. โคลนควรก้าวไปข้างหน้าเมื่อการแก้ไขที่ตั้งใจไว้ได้รับการยืนยันแล้ว, ไซต์ที่คาดว่าจะมีผลกระทบที่ไม่ตั้งใจชัดเจน, และบันทึกทั้งหมดอยู่ในที่แล้ว
การแก้ไขจีโนมด้วย CRISPR: วิธีลดผลกระทบที่ไม่ตั้งใจอย่างมีประสิทธิภาพ
บทสรุป: เช็คลิสต์สามขั้นตอนสำหรับการแก้ไขสายเซลล์ที่สะอาดขึ้น
เมื่อรวมกันแล้ว เช็คลิสต์นี้ถือว่าการควบคุมผลกระทบที่ไม่ตั้งใจเป็นกระบวนการที่มีขั้นตอน ไม่ใช่การตรวจสอบ QC เพียงครั้งเดียว เป้าหมายคือการลดความเสี่ยงตั้งแต่ต้น จำกัดกิจกรรมของตัวแก้ไข แล้วตรวจสอบผลลัพธ์
ความลึกของการตรวจสอบควรตรงกับความเสี่ยง ปล่อยเฉพาะโคลนที่เป็นอิสระหลายตัวที่ได้รับการยืนยันในสถานะอัลลีลที่ตั้งใจไว้เท่านั้น
คำถามที่พบบ่อย
ทำไมไม่พึ่งพาโคลนเดียว?
การพึ่งพาโคลนเดียวมีความเสี่ยง การแก้ไข CRISPR ไม่ได้มีความเฉพาะเจาะจงอย่างสมบูรณ์, ดังนั้นอาจทำให้เกิดการกลายพันธุ์นอกเป้าหมายที่ไม่ตั้งใจได้
นั่นคือเหตุผลที่ทีมงานมักจะขยายประชากรโคลนหลายตัว การทำเช่นนี้ทำให้ง่ายต่อการค้นหาเส้นที่มีการแก้ไขเป้าหมายที่ตั้งใจไว้โดยไม่มีการเปลี่ยนแปลงนอกเป้าหมายที่เป็นอันตราย
ยังมีเหตุผลอื่นด้วย: เซลล์ไลน์สามารถแสดงความหลากหลายทางพันธุกรรม การจัดลำดับโคลนหลายตัวช่วยยืนยันว่าการ น็อคเอาท์แบบโฮโมไซกัส หรือการดัดแปลงไซต์เป้าหมายอื่น ๆ มีอยู่ในตำแหน่งเป้าหมายทั้งหมด
เมื่อใดที่การทำแอมพลิคอน NGS เพียงพอ
การทำแอมพลิคอนโดยใช้เทคโนโลยีการหาลำดับเบสรุ่นใหม่มักจะเพียงพอเมื่อคุณต้องการวิธีที่มีเป้าหมายและคุ้มค่าในการยืนยันตำแหน่งที่อาจจะไม่ตรงเป้าหมายที่ถูกระบุโดยเครื่องมือคำนวณหรือวิธีการคัดกรองอื่น ๆ
การหาลำดับเบสทั้งจีโนมยังคงเป็นวิธีเดียวที่จะวัดผลกระทบที่ไม่ตรงเป้าหมายได้อย่างเต็มที่ แต่สำหรับการใช้งานหลาย ๆ อย่าง การวิเคราะห์ในระดับนั้นไม่จำเป็น
ฉันจะเลือกตัวแก้ไขที่ปลอดภัยที่สุดได้อย่างไร
เลือก ตัวแปรนิวเคลียส CRISPR ที่มีความเคลื่อนไหวน้อยที่สุด ที่ยังคงตัดตำแหน่งเป้าหมายของคุณได้ดี
คุณไม่สามารถเลือกตัวแปรที่ดีที่สุดจากการทำนายเพียงอย่างเดียว วิธีที่เชื่อถือได้เพียงวิธีเดียวคือการทำการคัดกรองขนาดเล็กในตัวแปรนิวเคลียสที่เลือกและอ่านผลการแก้ไขด้วย การหาลำดับเบสรุ่นใหม่.
สำหรับการวิจัยและพัฒนาของเนื้อสัตว์ที่เพาะเลี้ยง& นี่คือแนวทางปฏิบัติที่เป็นไปได้: เริ่มต้นด้วยรายการตัวแปรสั้น ๆ แล้วทดสอบตัวที่อ่อนแอกว่าทีละขั้นตอนจนกว่าคุณจะพบตัวเลือกที่มีความเคลื่อนไหวน้อยที่สุดที่ยังคงแก้ไขไซต์เป้าหมายได้อย่างมีประสิทธิภาพ.









