

In Reinräumen, stellt die GMP-Konformität durch detaillierte Überwachung und genaue Datenaufzeichnungen eine gleichbleibende Qualität und Sicherheit sicher. Für Anlagen zur Herstellung von kultiviertem Fleisch ist dies besonders wichtig, da selbst geringfügige Abweichungen in den Reinraumbedingungen das Zellwachstum beeinträchtigen oder Produktionschargen kontaminieren können.
Wichtige Erkenntnisse:
- GMP-Standards: Fokus auf Datenintegrität, gemäß dem ALCOA+-Rahmenwerk (Attributable, Lesbar, Zeitnah, Original, Genau, Vollständig, Konsistent, Dauerhaft, Verfügbar).
- Kritische Parameter: Überwachen Sie Luftpartikel, Keimzahlen, Temperatur, Luftfeuchtigkeit und Druck, um Risiken frühzeitig zu erkennen. Dies erfordert die Auswahl präziser Sensoren, die in der Lage sind, diese kritischen Parameter aufrechtzuerhalten.
- Datensysteme: Verwenden Sie validierte Bioprozesskontrollsysteme mit rollenbasierter Zugriffskontrolle, Prüfpfaden und sicherer Speicherung sowohl für elektronische als auch für Papieraufzeichnungen.
- Häufige Risiken: Vermeiden Sie Fehler bei der manuellen Datenverarbeitung, unkontrollierte Konfigurationsänderungen und unsachgemäße Lagerpraktiken.
- Maßgeschneiderte GMP für kultiviertes Fleisch: Passen Sie Überwachungsstrategien an, um einzigartige Risiken wie Bioreaktorbedingungen und Rückstände von Reinigungsmitteln zu adressieren.
Für kultiviertes Fleisch F&E, sorgt ein robustes Datenmanagement für Produktsicherheit, regulatorische Konformität und skalierbare Abläufe. Bekannte Schwachstellen proaktiv angehen, um später kostspielige regulatorische Probleme zu vermeiden.
Wichtige GMP-Anforderungen für die Datenintegrität im Reinraum
Verständnis der ALCOA+-Prinzipien
Der Grundstein der GMP-Datenintegrität liegt im ALCOA+-Rahmenwerk. Regulierungsbehörden wie die MHRA, EMA, und WHO verwenden es, um zu bestimmen, ob Reinraumaufzeichnungen vertrauenswürdig sind.ALCOA+ steht für: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, und Available. Jeder dieser Begriffe hat praktische Bedeutung in Reinraumoperationen.
- Attributable: Jeder Eintrag - sei es eine Partikelzählung, ein Druckwert oder ein Reinigungsprotokoll - muss klar zeigen, wer ihn aufgezeichnet hat, zusammen mit Datum, Uhrzeit und relevanten Instrumentendetails.
- Legible: Aufzeichnungen müssen leicht lesbar und verständlich sein, um Klarheit bei Überprüfungen oder Inspektionen zu gewährleisten.
- Contemporaneous: Daten müssen in Echtzeit erfasst werden. Verzögerte oder nachträgliche Einträge können die Zuverlässigkeit der Aufzeichnungen beeinträchtigen.
- Original: Daten sollten in ihrer ursprünglich erfassten Form bleiben, ohne unautorisierte Bearbeitungen oder Änderungen.
- Genau: Aufgezeichnete Werte müssen die beobachteten Ergebnisse wahrheitsgemäß widerspiegeln, frei von Fehlern oder Manipulationen.
- Vollständig: Alle relevanten Einträge, einschließlich Abweichungen oder Ergebnissen außerhalb der Spezifikation, müssen dokumentiert werden.
- Konsistent, Beständig, und Verfügbar: Aufzeichnungen sollten der richtigen Reihenfolge folgen, intakt für den erforderlichen Aufbewahrungszeitraum erhalten bleiben und leicht zugänglich für Überprüfung oder Inspektion sein.
Regulierungsbehörden legen großen Wert auf diese Prinzipien. Beispielsweise ergab eine MHRA-Inspektionsüberprüfung, dass über 80 % der GMP-Nichtkonformitätserklärungen in einem Zeitraum von drei Jahren Datenintegritätsprobleme betrafen [5]. Um ALCOA+ in den täglichen Arbeitsablauf zu integrieren, können Einrichtungen gut strukturierte Formulare einführen, Pflichtfelder durchsetzen und regelmäßige Überprüfungen der Audit-Trails durchführen.
Mit ALCOA+ als Grundlage besteht der nächste Schritt darin, sicherzustellen, dass diese Prinzipien in Papier-, elektronischen und hybriden Systemen eingehalten werden.
Sicherstellung der Datenintegrität über alle Formate hinweg
In Anlagen für kultiviertes Fleisch, wo Daten direkt die Entscheidungen zur Chargenfreigabe beeinflussen, ist die Aufrechterhaltung der Integrität über alle Aufzeichnungsformate hinweg unverzichtbar. GMP erfordert das gleiche Maß an Integrität für sowohl Papier- als auch elektronische Aufzeichnungen, obwohl die spezifischen Kontrollen je nach Format unterschiedlich sein können.
- Papiersysteme: Beste Praktiken umfassen die Verwendung von kontrollierten Formularen mit unauslöschlicher Tinte und klaren Korrekturprotokollen (e.g. , Korrekturen mit einem einzigen Durchstreichen, Unterschriften und Daten). Sichere Archivierung und Einhaltung definierter Aufbewahrungsfristen sind ebenfalls entscheidend.
- Elektronische Systeme: Diese sollten auf validierter Software basieren, die den Anforderungen von Anhang 11 und GAMP 5 . entspricht.Wichtige Funktionen umfassen rollenbasierter Zugriff, einzigartige Benutzer-Authentifizierung, umfassende Prüfpfade und synchronisierte Zeitstempel. Regelmäßige Überprüfungen der Prüfpfade sind unerlässlich, um Unregelmäßigkeiten zu identifizieren und zu beheben.
- Hybridsysteme: Diese stellen das höchste Risiko dar, da sie sowohl elektronische als auch Papieraufzeichnungen beinhalten. Zum Beispiel, wenn ein Instrument elektronische Daten erzeugt, die später in ein Papierprotokoll übertragen werden, muss die ursprüngliche elektronische Ausgabe als primäre Aufzeichnung erhalten bleiben. Abstimmungsschritte sollten in den Arbeitsablauf integriert werden, um Abweichungen zwischen den elektronischen und Papieraufzeichnungen zu erkennen und zu beheben. Dies ist besonders kritisch in der Produktion von kultiviertem Fleisch, wo selbst geringfügige Dateninkonsistenzen die Kontaminationskontrollmaßnahmen. gefährden könnten.
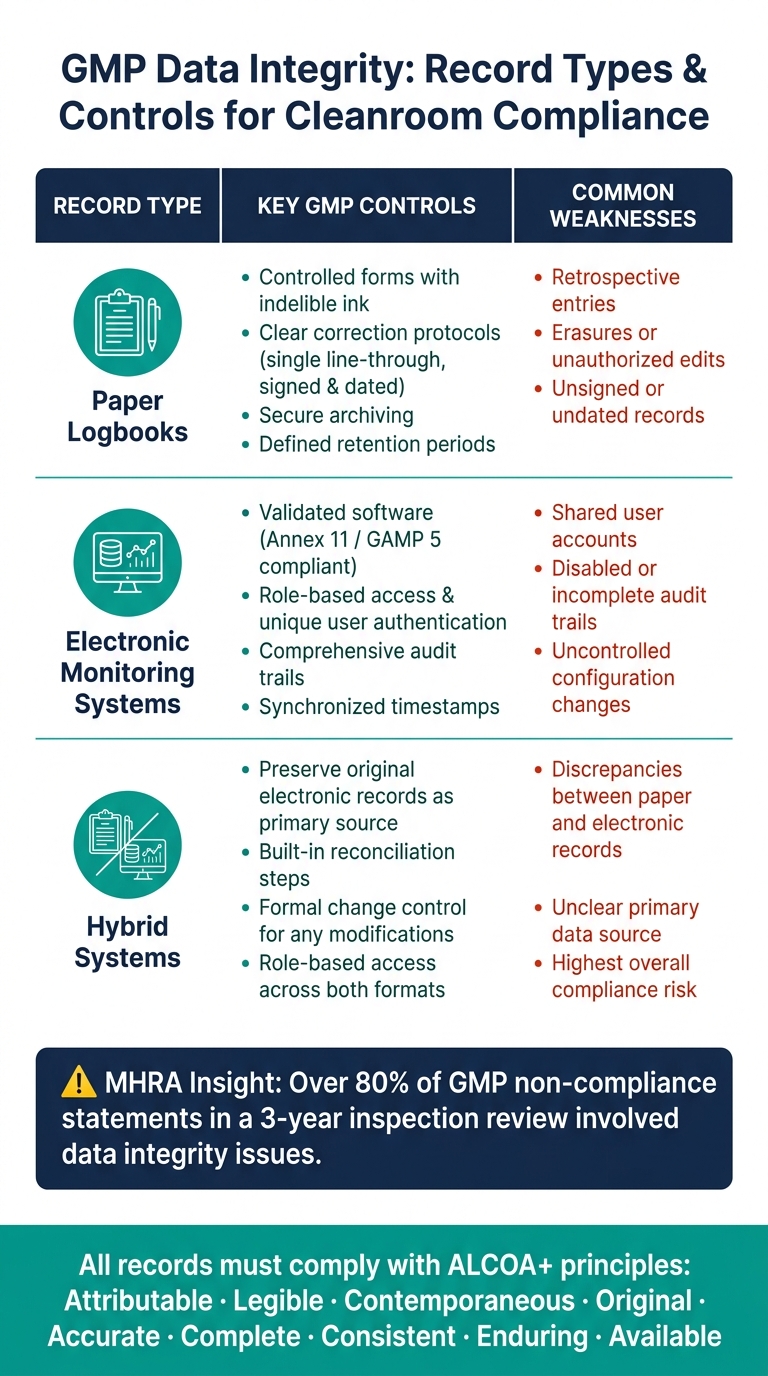
Die folgende Tabelle fasst die wichtigsten Kontrollen und häufigsten Schwächen für jeden Aufzeichnungstyp zusammen:
| Aufzeichnungstyp | Wichtige GMP-Kontrollen | Häufige Schwächen |
|---|---|---|
| Kontrollierte Formulare, unauslöschliche Tinte, klare Korrekturprotokolle, unterschriebene und datierte Einträge | Rückwirkende Einträge, Radierungen, nicht unterschriebene Aufzeichnungen | |
| Elektronische Überwachungssysteme | Validierte Software, rollenbasierter Zugriff, Prüfpfade, Zeitsynchronisation | Gemeinsame Benutzerkonten, deaktivierte oder unvollständige Prüfpfade |
| Hybridsysteme | Original elektronische Aufzeichnungen bewahren; Abstimmungsschritte implementieren | Abweichungen zwischen Papier- und elektronischen Aufzeichnungen, unklare primäre Datenquelle |
Um die Einhaltung sicherzustellen, sollten Aufzeichnungen nach ihrer Kritikalität kategorisiert werden.Für Anlagen zur Herstellung von kultiviertem Fleisch sollten Daten, die mit Chargenfreigabeentscheidungen oder Kontaminationskontrolle (e.g. , Ergebnisse der Umweltüberwachung, HVAC-Alarmprotokolle oder Filterintegritätstestdaten) verbunden sind, den strengsten Zugriffskontrollen, häufigen Überprüfungen und einem robusten Audit-Trail-Management unterliegen.
sbb-itb-ffee270
GMP-Reinraum Routine-Umweltüberwachung & 21CFR Teil 11 Datenintegrität
Verwaltung des Reinraum-Datenlebenszyklus
GMP-Datenintegrität: Aufzeichnungstypen & Kontrollen für Reinraum-Compliance
Phasen des Reinraum-Datenlebenszyklus
Der Reinraum-Datenlebenszyklus in Anlagen zur Herstellung von kultiviertem Fleisch umfasst mehrere Phasen, jede mit spezifischen Compliance-Anforderungen.
Datengenerierung markiert den Anfang. Dies umfasst Messungen von Instrumenten wie Partikelzählern, Differenzdrucksensoren, Temperatur- und Feuchtigkeitssonden, luftgängigen Probenahmegeräten, Oberflächenkontaktplatten und Reinigungsüberprüfungsprotokollen. Für jeden Parameter muss eine dokumentierte Probenahmefrequenz, ein zugewiesener Bediener und ein kalibriertes Instrument vorhanden sein. Die Ausrichtung dieser Überwachungsaufgaben auf Produktionsstufen - wie Inokulation, Zellexpansion oder Ernte - hilft zu demonstrieren, wie die Umweltkontrolle direkt mit der Produktqualität und -sicherheit verbunden ist.
Sobald die Daten generiert sind, gelangen sie in die Erfassungs- und Übertragungsphase. Idealerweise sollten elektronische Systeme Messwerte automatisch mit zeitgestempelten Einträgen aufzeichnen, die an individuelle Benutzerkonten gebunden sind. Bei papierbasierten Einträgen müssen die Daten in Echtzeit mit unauslöschlicher Tinte protokolliert werden, wobei bei der Übertragung der Daten in elektronische Systeme Abgleichsprüfungen durchgeführt werden.
Die Speicherphase ist ebenso kritisch.Sowohl Rohdaten als auch verarbeitete Daten müssen aufbewahrt werden, damit jeder gemeldete Wert auf seinen ursprünglichen Datensatz zurückverfolgt werden kann. Dies erfordert sichere, validierte Repositories mit rollenbasierten Zugriffskontrollen und regelmäßigen Backup-Tests. Backups sollten an einem separaten Ort vom primären System gespeichert und regelmäßig überprüft werden, um sicherzustellen, dass sie bei Bedarf wiederhergestellt werden können.
Schließlich schließt die Archivierung den Lebenszyklus ab. Datensätze wechseln in einen schreibgeschützten, kontrollierten Zugriffsstatus, sobald sie nicht mehr aktiv genutzt werden, müssen jedoch für den erforderlichen Aufbewahrungszeitraum abrufbar bleiben. In Anlagen für kultiviertes Fleisch kann die Archivierung von Daten aus der Entwicklungsphase auch zukünftige Validierungsbemühungen unterstützen.
Ein klares Verständnis dieser Phasen ist entscheidend für das effektive Risikomanagement, wie unten beschrieben.
Häufige Risiken im Datenmanagement
Die Datenverarbeitung während der Übertragungen birgt erhebliche Risiken.Manuelle Transkriptionsfehler und rückwirkende Einträge können die Datenintegrität untergraben. Um dies zu vermeiden, müssen alle Einträge den ALCOA+-Prinzipien (Attributable, Lesbar, Zeitnah, Original, Genau, plus Vollständigkeit, Konsistenz, Dauerhaft und Verfügbar) in Echtzeit entsprechen.
Konfigurationsänderungen sind ein weiteres großes Anliegen. Anpassungen der Alarmgrenzen, Sensorzuordnungen oder Systemeinstellungen ohne formale Änderungssteuerung können die Zuverlässigkeit der vor und nach der Änderung aufgezeichneten Daten beeinträchtigen. Zusätzlich können Speicherfehler - sei es durch beschädigte Datenbanken, ungetestete Backups oder durch Umwelteinflüsse beschädigte Papierarchive - kritische Aufzeichnungen unzugänglich machen. Um diese Risiken zu mindern, stellen Sie sicher, dass jeder Datenstrom einem bestimmten Archivierungspunkt mit klarer Zuständigkeit zugeordnet ist, um die Wahrscheinlichkeit zu verringern, dass Schwachstellen bei behördlichen Inspektionen aufgedeckt werden.
GMP-Kontrollen für Reinraum-Überwachungssysteme
Kritische Kontrollen für Überwachungssysteme
Ein Überwachungssystem, das den GMP-Standards entspricht, basiert auf kalibrierten Sensoren, sicherer Datenverarbeitung und effektivem Alarmmanagement. Sensoren für Parameter wie Temperatur, relative Luftfeuchtigkeit, Differenzdruck, nicht-lebensfähige Partikelzählung und lebensfähige mikrobiologische Probenahme müssen gemäß dokumentierten Zeitplänen kalibriert und auf anerkannte Standards rückführbar sein. Die Automatisierung der Datenübertragung von diesen Sensoren, komplett mit synchronisierten Zeitstempeln, minimiert das Risiko manueller Fehler.
Das Alarmmanagement ist ebenso entscheidend. Alarmgrenzen sollten mit regulatorischen Rahmenwerken wie ISO 14644-1 Klassengrenzen und EU GMP Anhang 1 Richtlinien übereinstimmen. Jeder ausgelöste Alarm muss von einer aufgezeichneten Reaktion begleitet werden, einschließlich Benutzerdetails, Zeitstempeln und eventuellen Kommentaren.Das Versäumnis, eine Alarmreaktion zu dokumentieren, schafft eine Compliance-Schwachstelle.
Rollenbasierte Zugriffskontrollen müssen im gesamten System strikt durchgesetzt werden. Für Änderungen an Alarmgrenzen, Sensorkonfigurationen oder Systemeinstellungen sollte eine Administrator-Autorisierung erforderlich sein, und diese Änderungen müssen einem formellen Änderungssteuerungsprozess folgen. Prüfpfade sind für alle GMP-bezogenen Aktionen obligatorisch, wie z.B. Konfigurationsaktualisierungen, Datenlöschungen, elektronische Signaturen und Sensoranpassungen. Diese Pfade müssen regelmäßig überprüft werden, wie in der MHRA-Datenintegritätsrichtlinie und dem EU-GMP-Anhang 11 beschrieben.
Für kultivierte Fleischproduktionssysteme, sind diese Kontrollen besonders wichtig, da Umweltbedingungen einen direkten Einfluss auf die Zellviabilität haben.
Sobald diese Kontrollen implementiert sind, muss das System validiert und Änderungen sorgfältig verwaltet werden, um die fortlaufende Compliance sicherzustellen.
Validierungs- und Änderungssteuerungsverfahren
Überwachungssysteme müssen durch die Phasen IQ, OQ und PQ validiert werden, um die Genauigkeit der Sensoren, die Funktionalität der Alarme, die Datenintegrität, die Backup-Prozesse und die Prüfpfade zu überprüfen. Alternativ kann ein Lebenszyklusansatz verwendet werden, der mit den Prinzipien von GAMP 5 und Anhang 11 übereinstimmt.
Der EU-GMP-Anhang 1 (Revision 2022) verlangt, dass Umweltsysteme "angemessen qualifiziert und validiert" sind und dass elektronische Aufzeichnungen den Standards von Anhang 11 für Integrität, Sicherheit und Rückverfolgbarkeit entsprechen. Diese Anforderungen bilden die Grundlage für jede GMP-konforme Einrichtung.
Alle Änderungen, die die Datenintegrität, die Funktionalität der Alarme oder die Rückverfolgbarkeit beeinflussen könnten, müssen einem formellen Änderungssteuerungsprozess unterzogen werden. Selbst scheinbar geringfügige Aktualisierungen, wie Software-Patches, können Prüfpfade stören und sollten nicht ohne vorherige Auswirkungenseinschätzung implementiert werden.
Verschiedene Datentypen erfordern maßgeschneiderte GMP-Kontrollen, um eine genaue und rechtzeitige Berichterstattung zu gewährleisten.
Vergleich von Datentypen und Compliance-Anforderungen
Jeder Datentyp in der Reinraumüberwachung hat spezifische Anforderungen, um die GMP-Compliance aufrechtzuerhalten.Die folgende Tabelle zeigt die wichtigsten Kontrollen für verschiedene Datentypen:
| Datentyp | Überwachungsmodus | Wichtige GMP-Kontrollen | Grenzbasis |
|---|---|---|---|
| Nicht-lebensfähige Partikelzählungen | Kontinuierlich oder häufig während der Operationen in Klasse A/B; routinemäßig in anderen Klassen | Validierte Partikelzähler; automatische Datenerfassung; alarmiert; Kalibrierung rückverfolgbar zu Standards; Audit-Trail für Konfigurationsänderungen | ISO 14644-1 Klassengrenzen; Anhang 1 Leitlinien für Klasse A/B Zonen |
| Differenzdruck | Kontinuierlich; alarmiert | Kalibrierte Drucktransmitter; automatische, zeitgestempelte Aufzeichnung; protokollierte Alarmbestätigungen; Aufrechterhaltung eines Differenzdrucks von 10–15 Pa zwischen den Zonen | Anhang 1; einrichtungsspezifisches Raumklassifikationsdesign |
| Temperatur und relative Luftfeuchtigkeit | Kontinuierlich für kritische Prozesse; periodisch an anderen Stellen | Kalibrierte Sonden; automatische Datenerfassung; Trendanalyse; Alarmgrenzen basierend auf Prozess- und regulatorischen Anforderungen | Prozesskenntnisse; regulatorische Richtlinien; Produktsensitivität |
| Lebensfähige luftgetragene Mikroben | Intermittierend (aktive Luftprobenahme); erhöhte Frequenz für kritische Operationen | Qualifizierte Probennehmer; kontrollierte Probenahmeverfahren; Nachverfolgung zur Labor; Ergebnisse verknüpft mit Charge und Standort; untersuchungsbereite Aufzeichnungen | EU-GMP-Anhang 1 mikrobiologische Grenzwerte nach Klasse |
| Oberflächenkontakt-Ergebnisse | Periodisch; nach Reinigung und nach Betrieb | Kontrollierte Probenahmemethoden; Rückverfolgbarkeit im Labor; Ergebnisse werden mit spezifischen Grenzwerten verglichen; verknüpft mit Reinigungsaufzeichnungen | EU GMP Anhang 1; Betriebs-SOPs |
Jeder Datentyp erfordert definierte Akzeptanzkriterien, regelmäßige Überprüfungspläne, Aufbewahrungsrichtlinien und einen Eskalationsprozess für Abweichungen.Die Anwendung einheitlicher Überprüfungsstandards auf alle Datentypen ist ein häufiger Fehler, den Regulierungsbehörden zunehmend unter die Lupe nehmen. Die Anpassung des Überprüfungsprozesses an die spezifischen Bedürfnisse jedes Datentyps gewährleistet Compliance und betriebliche Effizienz.
Berichterstattung, Überprüfung und Korrekturmaßnahmen
Erstellung konformer Berichte
Berichte, die den ALCOA+-Standards entsprechen, müssen gründlich, präzise und für Audits zugänglich sein. Ein GMP-konformer Reinraumüberwachungsbericht sollte prägnant, überprüfbar und in der Lage sein, Entscheidungen zur Chargenfreigabe zu unterstützen, während er die Umweltkontrolle demonstriert. Diese Berichte sollten mindestens:
- Den Überwachungszeitraum und -umfang abdecken.
- Die Probenahmeaktivitäten im Vergleich zum geplanten Zeitplan zusammenfassen.
- Klar angeben, ob Warn- oder Aktionsgrenzen überschritten wurden.
Trendanalysen sind ein wesentlicher Bestandteil dieser Berichte und verwenden statistische Werkzeuge wie Regelkarten, gleitende Durchschnitte und Exkursionsraten pro 100 Proben, um allmähliche Veränderungen zu identifizieren. Beispielsweise bietet ein monatlicher Trend, der einen stetigen Anstieg der lebensfähigen Zählungen in der Nähe einer Bioreaktor-Erntelinie zeigt, weit mehr Einblick als ein einzelnes Ereignis außerhalb der Grenzwerte. Das Hinzufügen von Anmerkungen, wie Wartungsaktivitäten, Prozessanpassungen oder Personaländerungen, macht die Daten leichter interpretierbar und auditbereit.
Audit-Trail-Überprüfungen sind ein weiterer kritischer Schritt, der geschultes Personal erfordert, um ihre Ergebnisse sorgfältig zu dokumentieren. Dies umfasst die Aufzeichnung, wer bestimmte Systemereignisse überprüft hat, das Notieren von Anomalien und die detaillierte Beschreibung von Folgemaßnahmen, alles innerhalb eines unterschriebenen und datierten Protokolls.
Die Berichtsfrequenz sollte mit dem verbundenen Risiko übereinstimmen. Zum Beispiel:
- Chargenbezogene Berichte werden für jeden Produktionslauf erstellt.
- Zusammenfassungen der routinemäßigen Umweltüberwachung sind typischerweise wöchentlich oder monatlich.
- Trendberichte werden monatlich oder vierteljährlich erstellt, um frühe Anzeichen von Abweichungen zu erkennen.
Die gewählte Berichterstattungshäufigkeit muss in Standardarbeitsanweisungen (SOPs) gerechtfertigt und konsequent eingehalten werden. Diese Protokolle bilden auch die Grundlage für die Einleitung von Korrekturmaßnahmen, wenn Abweichungen festgestellt werden.
Behebung von Compliance-Verstößen
Wenn Abweichungen auftreten, ist eine strukturierte, nachvollziehbare Reaktion unerlässlich. Jede Abweichung sollte eine eindeutige Kennung, eine klare Beschreibung und eine Risikobewertung haben, die sowohl die Produktwirkung als auch die Datenintegrität bewertet. Abweichungen müssen auch klassifiziert werden (geringfügig, bedeutend oder kritisch) und mit bestimmten Chargen oder Produktionsläufen verknüpft werden, um zu beurteilen, ob die Chargenfreigabe betroffen ist oder zusätzliche Tests erforderlich sind.
Der CAPA (Corrective and Preventive Action) Rahmen ist zentral für die Behebung von GMP-Verstößen. Effektive CAPA erfordert mehr als die Zuschreibung von Ereignissen auf "menschliches Versagen." EMA und PIC/S Leitlinien betonen:
"das Versäumnis, kritische Abweichungen, OOS-Ergebnisse und Datenintegritätsprobleme angemessen zu untersuchen" ist eine wiederkehrende Ursache für Durchsetzungsmaßnahmen.
Werkzeuge zur Ursachenanalyse, wie die 5 Whys oder Fischgrätendiagramme, sind von unschätzbarem Wert, um systemische Probleme aufzudecken - sei es in Bezug auf Verfahrenslücken, unzureichende Schulung oder Schwächen in technischen Kontrollen. Korrekturmaßnahmen sollten die während der Datenerfassung und -speicherung identifizierten Risiken adressieren.
Jede CAPA muss messbare Wirksamkeitskriterien enthalten. Zum Beispiel: "keine wiederholten Grade B lebensfähigen Überschreitungen über dem Aktionsniveau für sechs Monate." Darüber hinaus sind Nachprüfungen unerlässlich, um sicherzustellen, dass diese Kriterien erfüllt werden. Häufige CAPA-Kennzahlen umfassen:
- Die Anzahl der offenen Maßnahmen.
- Durchschnittliche Zeit bis zum Abschluss.
- Prozentsatz der pünktlich abgeschlossenen Maßnahmen.
- Rate der wiederholten Abweichungen, die als starker Indikator für die Wirksamkeit von CAPA dient.
Eine Überprüfung der GMP-Warnschreiben von 2015 bis 2019 ergab, dass 65–70 % der Datenintegritätszitate auf unzureichende Untersuchungen, fehlende Dokumentation oder Versäumnisse bei der ordnungsgemäßen Überprüfung und Berichterstattung von Daten zurückzuführen waren [2]. Dies unterstreicht die Bedeutung einer robusten Berichterstattung und eines reaktionsfähigen CAPA-Rahmens als Nachweis für eine gut kontrollierte Einrichtung.
Aufrechterhaltung der GMP-Konformität in kultivierten Fleischanlagen
Um Sicherheit und Qualität in der Produktion von kultiviertem Fleisch zu gewährleisten, müssen Einrichtungen etablierte GMP-Kontrollen anpassen, um den spezifischen Herausforderungen dieses aufstrebenden Bereichs gerecht zu werden.Da die Datenintegrität im Reinraum eine entscheidende Rolle bei der Aufrechterhaltung der Produktsicherheit spielt, ist die Verfeinerung der GMP-Praktiken für kultiviertes Fleisch unerlässlich.
Anpassung der GMP-Praktiken für kultiviertes Fleisch
GMP-Rahmenwerke wie EU-Anhang 1, ursprünglich für Pharmazeutika erstellt, erfordern Anpassungen, um die einzigartigen Risiken in der Produktion von kultiviertem Fleisch zu adressieren. Eine formale Risikoanalyse, wie eine FMEA oder HACCP-ähnliche Analyse, bietet eine solide Grundlage, um GMP-Prinzipien mit jeder Produktionsstufe in Einklang zu bringen. Kritische Operationen wie das Auftauen von Zellbanken, die Inokulation von Bioreaktoren, die Zellexpansion und die Ernte erfordern geeignete Reinraumklassifikationen, Bekleidungsprotokolle und Umweltüberwachung, wie in Anhang 1 spezifiziert.In der Zwischenzeit können nachgelagerte Aufgaben wie Gerüsthandhabung und Verpackung den lebensmittelgerechten Hygiene-GMP-Standards gemäß Verordnung (EG) Nr. 852/2004, entsprechen, vorausgesetzt, die Rückverfolgbarkeit und Datenintegrität bleiben während des gesamten Prozesses intakt [6] [9][14].
Umweltüberwachungsstrategien sollten sich auf Organismen konzentrieren, die für kultiviertes Fleisch und Lebensmittelsicherheit relevant sind, anstatt ausschließlich traditionelle pharmazeutische Krankheitserreger ins Visier zu nehmen. Probenahmen sollten in Hochrisikobereichen priorisiert werden, wie z. B. in der Nähe von offenen Bioreaktoren, Medienvorbereitungszonen und Gerüsthandhabungsstationen. Diese Standorte sollten basierend auf dokumentierten Luftströmungsmustern und Analysen der Personalbewegungen ausgewählt werden [9][10].
Angesichts des hohen Datenvolumens, das von Bioreaktoren für kultiviertes Fleisch erzeugt wird, müssen Systeme in der Lage sein, diese Daten zu erfassen, mit Zeitstempeln zu versehen und sicher in einem validierten Repository zu speichern. Die ursprüngliche Rohdatendatei des Instruments sollte immer als Hauptaufzeichnung identifiziert werden, um die Einhaltung sicherzustellen [7][8].
Reinigungs- und Desinfektionsprotokolle erfordern ebenfalls sorgfältige Überlegungen. Rückstände, die in pharmazeutischen Umgebungen als akzeptabel gelten, könnten die Zelladhäsion oder -differenzierung in der Produktion von kultiviertem Fleisch beeinträchtigen. Verifizierungsdaten für Reinigungsmittel sollten gesammelt und als Teil des Umweltüberwachungsprogramms aufbewahrt werden [3][4].
Verwendung von Branchenressourcen wie Cellbase
Spezialisierte Beschaffungsplattformen sind von unschätzbarem Wert, um die spezifischen Bedürfnisse von Anlagen für kultiviertes Fleisch zu erfüllen. GMP-kompatible Reinraumausrüstung sollte den Schutz- und Reinigungsstandards entsprechen und sich nahtlos in validierte Datensysteme integrieren. Lieferanten müssen detaillierte Spezifikationen bereitstellen, einschließlich Audit-Trail-Funktionen, Datenexportformate, Alarmkonfigurationen und Kalibrierungsverfahren, zusammen mit der Ausrüstung.
Bei der Beschaffung von Systemen über
- Datenintegritätsfunktionen: sichere Audit-Trails, zeitgestempelte Aufzeichnungen, rollenbasierte Berechtigungen und eindeutige Benutzeranmeldungen
- Systemkompatibilität: Unterstützung für standardisierte Kommunikationsprotokolle und APIs für zentrale Datenspeicherung
- Kalibrierung und Wartung: Verfügbarkeit umfassender Dokumentation
- Qualifikationsunterstützung: vom Lieferanten bereitgestellte IQ/OQ-Vorlagen für eine vereinfachte Validierung
- Reinraumtauglichkeit: Materialien und Designs, die die Reinigung erleichtern
Das frühzeitige Anfordern von Lieferantendokumentationen im Beschaffungsprozess kann helfen, potenzielle Qualifikationsprobleme zu vermeiden [3][4].
Fazit
Zusammenfassung der wichtigsten Punkte
Die GMP-Konformität im Reinraum-Datenmanagement dreht sich um die Demonstration der Kontrolle - über Prozesse, Aufzeichnungen und die durch diese Daten informierten Entscheidungen. Wenn eine Aufzeichnung an Zuverlässigkeit mangelt, wird der dokumentierte Prozess ebenso fragwürdig. Dieses Prinzip gilt universell, sei es bei der Überwachung von Umweltprotokollen, Bioreaktor-Ausgaben, Abweichungsberichten oder Kalibrierungszertifikaten.
Vier zentrale Themen haben sich im Laufe dieser Diskussion herauskristallisiert. Erstens, Datenintegrität, geleitet von den ALCOA+-Prinzipien, ist der Grundstein für konforme Reinraumdokumentation. Zweitens, Lebenszyklusmanagement stellt sicher, dass Daten genau aufgezeichnet, umgehend überprüft, sicher gespeichert und für die erforderliche Dauer aufbewahrt werden. Drittens, validierte und änderungskontrollierte Überwachungssysteme bilden die technische Grundlage, die kein SOP ersetzen kann.Wie die Analyse der GMP-Inspektionen der MHRA von 2016 bis 2021 zeigt, gehören unvollständige Aufzeichnungen und unzureichende Überprüfungen der Audit-Trails weiterhin zu den häufigsten Mängeln. Schließlich stellt eine genaue und nachvollziehbare Berichterstattung sicher, dass Rohdaten mit Chargenentscheidungen, Untersuchungen und Korrekturmaßnahmen verknüpft werden können, um den regulatorischen Erwartungen zu entsprechen. Für Anlagen zur Herstellung von kultiviertem Fleisch gewinnen diese Prinzipien noch mehr an Bedeutung. Die Herausforderung, R&D-ähnliche Arbeitsabläufe mit Produktionskontrollen zu kombinieren, erfordert eine robuste Datenverwaltung, um beide Betriebsumgebungen zu überbrücken. Eine ordnungsgemäße Datenverwaltung im Reinraum gewährleistet nicht nur Konsistenz und Reproduzierbarkeit, sondern bereitet die Anlagen auch auf die Skalierung vor und demonstriert die Produktsicherheit gegenüber Regulierungsbehörden, Investoren und Verbrauchern. Der praktischste Rat? Bekannte Risiken angehen, bevor Prüfer sie hervorheben.Schwachstellen wie hybride Papier-Elektronik-Systeme, geteilte Benutzeranmeldungen, verzögerte Datenüberprüfungen und unkontrollierte lokale Speicherung sind vorhersehbar und vermeidbar. Die proaktive Lösung dieser Probleme ist weitaus effektiver - und kostengünstiger - als die Rekonstruktion einer Datenverfolgung nach einem Qualitätsvorfall.
Für Teams, die Überwachungsgeräte, Sensoren oder Infrastrukturen suchen, die auf diese Bedürfnisse zugeschnitten sind,
FAQs
Wie kann ALCOA+ in täglichen Reinraumaufzeichnungen nachgewiesen werden?
Um ALCOA+-Prinzipien in täglichen Reinraumaufzeichnungen anzuwenden, stellen Sie Folgendes sicher:
- Zurechenbar: Identifizieren Sie klar die verantwortliche Person, einschließlich Zeitstempel für jeden Eintrag.
- Lesbar: Aufzeichnungen müssen leicht lesbar und frei von Mehrdeutigkeiten sein.
- Gleichzeitig: Dokumentieren Sie Informationen zum Zeitpunkt der Aktivität.
- Original: Bewahren Sie die erste Aufzeichnung von Daten auf, keine Kopien oder Abschriften.
- Genau: Stellen Sie sicher, dass alle Einträge die tatsächlichen Daten fehlerfrei widerspiegeln.
- Vollständig: Schließen Sie alle relevanten Daten und Metadaten ohne Auslassungen ein.
- Konsistent: Bewahren Sie eine logische, sequentielle Reihenfolge in den Aufzeichnungen.
- Dauerhaft: Verwenden Sie Formate und Materialien, die für die Langzeitarchivierung geeignet sind.
- Verfügbar: Halten Sie Aufzeichnungen für Überprüfungen oder Audits bei Bedarf zugänglich.
Diese Schritte sind entscheidend, um die Einhaltung der Guten Herstellungspraxis (GMP) im Reinraum-Datenmanagement sicherzustellen.
Was sind die Hauptdatenintegritätsrisiken in hybriden Papier-Elektronik-Systemen?
Die Speicherung von Informationen an mehreren Standorten führt zu Komplikationen bei der Überprüfung der Datengenauigkeit. Darüber hinaus erhöht die manuelle Dateneingabe das Risiko menschlicher Fehler, während schlecht kontrollierte oder eigenständige Systeme Aufzeichnungen anfällig für Manipulation oder Löschung machen. Diese Probleme unterstreichen die Notwendigkeit starker Datenmanagementpraktiken, um die Einhaltung der Vorschriften zu gewährleisten und die Datenintegrität zu bewahren.
Welche Nachweise erwarten Inspektoren für die Überwachung der Systemvalidierung und Änderungssteuerung?
Inspektoren fragen oft nach dokumentierten Nachweisen, die die Systemvalidierung zeigen. Dazu gehört das Testen kritischer Parameter wie:
- HEPA-Filter-Integrität: Sicherstellen, dass die Filter die erforderlichen Leistungsstandards erfüllen.
- Luftstrom und Druckdifferenzen: Überprüfung, dass diese innerhalb akzeptabler Bereiche liegen, um kontrollierte Umgebungen aufrechtzuerhalten.
- Umweltdatenüberwachung: Nachweis, dass die Einrichtung die Anforderungen an Sauberkeit und Kontaminationskontrolle erfüllt.
Über die Validierungstests hinaus ist die Führung von Aufzeichnungen über Änderungssteuerungsaktivitäten ebenso wichtig. Dies umfasst Maßnahmen wie Filterwechsel oder Einrichtungsänderungen, die dazu beitragen, nachzuweisen, dass das System weiterhin wie erwartet funktioniert und den behördlichen Standards entspricht.










