

Die Reinraumvalidierung stellt sicher, dass Produktionsumgebungen strenge Kontaminationsstandards erfüllen, was für die Produktion von kultiviertem Fleisch entscheidend ist. Dies ist ein kritischer Schritt beim Hochskalieren von kultivierten Fleischprozessen. Eine ordnungsgemäße Validierung verhindert Kontaminationsrisiken, schützt die Produktqualität und erfüllt Vorschriften wie ISO 14644 und GMP. Der Prozess umfasst vier Schlüsselphasen:
- Designqualifikation (DQ): Bestätigt, dass das Reinraumdesign den betrieblichen und regulatorischen Anforderungen entspricht.
- Installationsqualifikation (IQ): Überprüft, ob die Komponenten korrekt installiert sind und den Spezifikationen entsprechen.
- Betriebsqualifikation (OQ): Testet Systeme im inaktiven Zustand, um sicherzustellen, dass sie wie vorgesehen funktionieren.
- Leistungsqualifikation (PQ): Bewertet die Reinraumleistung während der tatsächlichen Produktion.
Testprotokolle, einschließlich Partikelzählungen, HEPA-Filter-Integritätsprüfungen und Luftstrommessungen, sind entscheidend für die Einhaltung der Vorschriften. Kontinuierliche Überwachung und regelmäßige Revalidierung tragen dazu bei, die Leistung des Reinraums im Laufe der Zeit aufrechtzuerhalten. Die Einhaltung dieser Schritte stellt sicher, dass Kontaminationsrisiken minimiert werden, was sowohl die Produktkonsistenz als auch die behördliche Zulassung schützt.
Reinraumvalidierung von URS bis PQ
sbb-itb-ffee270
Die 4 Phasen der Reinraumvalidierung
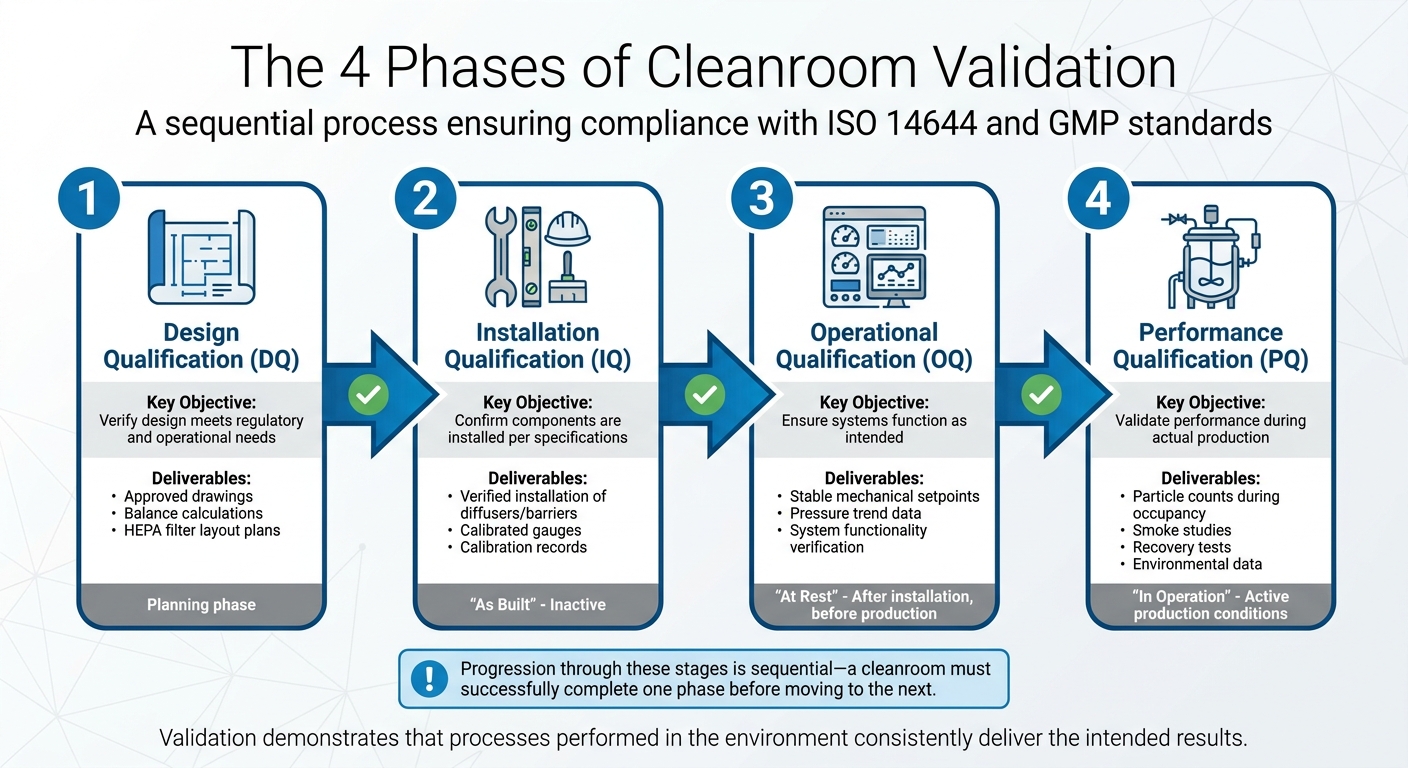
4 Phasen der Reinraumvalidierung für die Produktion von kultiviertem Fleisch
Die Reinraumvalidierung ist ein schrittweiser Prozess mit vier unterschiedlichen Phasen, die jeweils auf der vorherigen aufbauen. Der Fortschritt durch diese Phasen erfolgt nacheinander - ein Reinraum muss eine Phase erfolgreich abschließen, bevor er zur nächsten übergehen kann.Wie Allied Cleanrooms treffend feststellt:
"Validierung ist das, was einen sauber aussehenden Reinraum von einem tatsächlich einsatzbereiten unterscheidet" [8].
Während die Qualifizierung sicherstellt, dass der Reinraum und seine Systeme wie geplant installiert und funktionieren, geht die Validierung einen Schritt weiter. Sie zeigt, dass die in der Umgebung durchgeführten Prozesse konsequent die beabsichtigten Ergebnisse liefern [7]. Die vier Phasen - Design Qualification (DQ), Installation Qualification (IQ), Operational Qualification (OQ) und Performance Qualification (PQ) - sind darauf ausgelegt, Einrichtungen für validierte Produktionsprozesse vorzubereiten. Diese Phasen legen auch den Grundstein für strenge Testprotokolle.
| Validierungsphase | Hauptziele | Typische Ergebnisse/Tests |
|---|---|---|
| Designqualifikation (DQ) | Überprüfen, ob das Design den regulatorischen und betrieblichen Anforderungen entspricht. | Genehmigte Zeichnungen, Bilanzberechnungen, HEPA-Filter-Layoutpläne. |
| Installationsqualifikation (IQ) | Bestätigen, dass die Komponenten gemäß den Spezifikationen installiert sind. | Verifizierte Installation von Diffusoren/Barrieren, kalibrierte Messgeräte. |
| Betriebsqualifikation (OQ) | Sicherstellen, dass Systeme wie vorgesehen funktionieren. | Stabile mechanische Sollwerte, Drucktrenddaten. |
| Leistungsqualifikation (PQ) | Leistung während der Produktion/Belegung validieren. | Partikelzählungen, Rauchstudien, Wiederherstellungstests, Umweltüberwachungsdaten. |
Designqualifikation (DQ)
Die Designqualifikationsphase (DQ) stellt sicher, dass das Design des Reinraums den spezifischen Anforderungen der Produktion von kultiviertem Fleisch entspricht. Dies beinhaltet die Überprüfung, dass Entwurfsdokumente, wie z.B. Bilanzberechnungen und HEPA-Filterlayouts, die tatsächlichen Betriebsanforderungen widerspiegeln. Jedes Designelement muss strenge Akzeptanzkriterien erfüllen, die oft auf ISO 14644-Standards oder benutzerdefinierten Anforderungen basieren [7].
Installationsqualifikation (IQ)
Die Installationsqualifikation (IQ) konzentriert sich darauf, den "wie gebaut"-Zustand des Reinraums in seinem inaktiven Zustand zu überprüfen. Diese Phase bestätigt, dass Diffusoren, Rückläufe und Barrieren den Designvorgaben entsprechen. Es wird auch überprüft, ob Druckmonitore und Messgeräte korrekt kalibriert und voll funktionsfähig sind.Detaillierte Dokumentation, einschließlich Kalibrierungsaufzeichnungen und kartierten Teststandorten, ist in dieser Phase entscheidend [7][8].
Betriebsqualifikation (OQ)
Die Betriebsqualifikation (OQ) testet den Reinraum im "Ruhezustand" - nach der Installation, aber bevor die Produktion beginnt. Diese Phase stellt sicher, dass die Systeme wie vorgesehen funktionieren, indem stabile mechanische Sollwerte und konsistente Drucktrends dokumentiert werden. Wenn wesentliche Änderungen auftreten, wie z.B. die Verlagerung von Geräten oder die Änderung des Luftstroms, sind gezielte Nachtests erforderlich, um das Gleichgewicht zu halten [7][8]. Sobald bestätigt ist, dass die Systeme korrekt arbeiten, ist die Einrichtung bereit für die Leistungsvalidierung unter aktiven Bedingungen.
Leistungsqualifikation (PQ)
Die letzte Phase, die Leistungsqualifikation (PQ), validiert die Leistung des Reinraums unter tatsächlichen Produktionsbedingungen.Diese Phase bewertet, ob die Einrichtung die Leistungsziele während der Nutzung für die Produktion von kultiviertem Fleisch erfüllt. Wichtige Bewertungen umfassen Partikelzählungen während der Belegung, Luftstromvisualisierung (wie Rauchstudien) um kritische Bereiche und Wiederherstellungstests, um zu messen, wie schnell der Raum nach einer Störung die erforderliche Sauberkeit wiedererlangt. Bevor mit PQ begonnen wird, stellen Sie sicher, dass die mechanischen Sollwerte stabil sind, verwaltet über Bioprozess-Steuerungssoftware, kritische Probenahmestellen identifiziert sind und Reinigungsaufzeichnungen validierte Bedingungen bestätigen [7].
Für Einrichtungen zur Produktion von kultiviertem Fleisch wird die Nutzung unabhängiger Validierungsagenturen von Drittanbietern dringend empfohlen. Diese unparteiische Überprüfung hat bei Regulierungsbehörden und Prüfern mehr Gewicht. Allied Cleanrooms betont:
"Regulierungsbehörden und Prüfer legen mehr Wert auf Ergebnisse, die von einer externen Partei ohne Eigeninteresse am Ergebnis stammen" [8].
Dieser unabhängige Ansatz ist besonders wichtig für Einrichtungen, die eine USDA-Inspektionsgenehmigung anstreben, die den erfolgreichen Abschluss der FDA-Vorkonsultation erfordert [5] [6].
Erforderliche Testprotokolle für die Reinraumvalidierung
Sobald die Designqualifikation (DQ), Installationsqualifikation (IQ), Betriebsqualifikation (OQ) und Leistungsqualifikation (PQ) abgeschlossen sind, folgt eine gründliche Reihe von Tests, um die Leistung des Reinraums zu überprüfen. Diese Tests stellen sicher, dass der Reinraum seiner ISO-Klassifizierung entspricht und für die Produktion von kultiviertem Fleisch geeignet ist. Nachfolgend finden Sie einen Überblick über die wichtigsten Testprotokolle.
Luftgetragene Partikelzähltests
Dieser Test misst die Anzahl der Partikel in der Luft, um zu bestätigen, dass der Reinraum seiner ISO-Klassifizierung entspricht.Zum Beispiel sollte ein ISO 5-Reinraum nicht mehr als 3.520 Partikel von 0,5 µm oder größer pro Kubikmeter überschreiten. Die Prüfung erfolgt mit kalibrierten Partikelzählern an festgelegten Probenahmestellen unter den Bedingungen "im Ruhezustand" und "im Betrieb". Gemäß ISO 14644-2 sollten Partikelkonzentrationstests alle sechs Monate für ISO 5 und strengere Klassifikationen und jährlich für ISO 6 und höher durchgeführt werden [8].
HEPA-Filter-Integritätstests
Diese Tests stellen sicher, dass Hochleistungs-Partikelluftfilter (HEPA) ordnungsgemäß funktionieren, ohne Lecks oder Defekte. Während Partikelzähltests die allgemeine Sauberkeit des Raums bewerten, konzentrieren sich Integritätstests auf die Filter selbst. Jede wesentliche Änderung, wie Filterwechsel oder Raumänderungen, erfordert sofortige Nachtests.Viele Einrichtungen entscheiden sich für Drittanbieter-Agenturen, um diese Tests durchzuführen, da unabhängige Überprüfungen von Regulierungsbehörden oft hoch geschätzt werden [8].
Luftstromgeschwindigkeit und Volumenmessungen
Ein ordnungsgemäßer Luftstrom ist entscheidend für die Aufrechterhaltung der Sauberkeit. Der Luftstrom in unidirektionalen Reinräumen sollte typischerweise innerhalb von 0,45 m/s ±20% (zwischen 0,36 und 0,54 m/s) liegen. Messungen werden normalerweise in Arbeitshöhe durchgeführt - dort, wo empfindliche Vorgänge wie die Inokulation von Bioreaktoren innerhalb skalierbarer Bioreaktorsysteme stattfinden - oder 150 bis 300 mm von der Filterfläche entfernt. ISO 14644-3:2005 legt fest, dass die Anzahl der Probenahmepunkte der Quadratwurzel aus dem Zehnfachen der Raumfläche (in Quadratmetern) entsprechen sollte, mit mindestens vier Messungen und mindestens einem Punkt pro Filter. Rauchstudien oder Strömungsvisualisierungskartierungen können den unidirektionalen Luftstrom weiter überprüfen und Bereiche mit stagnierender Luft, bekannt als "Nachlaufzonen", erkennen [9] .
Druckdifferenzprüfungen
Die Aufrechterhaltung ordnungsgemäßer Druckdifferenzen zwischen Reinraumbereichen ist entscheidend, um Kontaminationen zu verhindern. Sauberere Bereiche müssen einen positiven Druck im Vergleich zu angrenzenden, weniger sauberen Bereichen aufrechterhalten. Kalibrierte Druckmessgeräte und Sensoren werden verwendet, um stabile Druckdifferenzen zu dokumentieren und sicherzustellen.
Temperatur- und Feuchtigkeitsüberprüfung
Die Temperatur- und Feuchtigkeitswerte des Reinraums müssen sorgfältig kontrolliert werden, um die Produktion von kultiviertem Fleisch zu unterstützen. Diese Bedingungen beeinflussen sowohl die Produktqualität als auch die Leistung von HEPA-Filtern und anderen Systemen. Eine kontinuierliche Überwachung hilft sicherzustellen, dass diese Parameter während der gesamten Produktionszyklen innerhalb der erforderlichen Sollwerte bleiben.
Kontinuierliche Überwachung und Revalidierung
Die Validierung endet nicht, sobald Systeme implementiert sind. Kontinuierliche Überwachung und regelmäßige Revalidierung sind entscheidend, um den Auswirkungen von Filterverschleiß, Verschlechterung des HLK-Systems und Prozessänderungen entgegenzuwirken. Nach Erreichen der anfänglichen Konformität durch DQ, IQ, OQ und PQ erfordert die Aufrechterhaltung der Leistung während der aktiven Produktion eine fortlaufende Überwachung.
Programme zur Umweltüberwachung
Ein robustes Umweltüberwachungsprogramm verfolgt die Anzahl der luftgetragenen Partikel, mikrobiologische Kontamination, Temperatur, Luftfeuchtigkeit und Druckdifferenzen gemäß einem definierten Zeitplan. In Grade-A-Zonen muss die Überwachung kontinuierlich erfolgen, während in Grade-B-Zonen alle 15–30 Minuten Kontrollen erforderlich sind. Grade-C- und D-Zonen können stündlich oder pro Schicht überwacht werden, basierend auf Risikobewertungen [3][4].
Mikrobielles Monitoring kombiniert aktive Luftprobenahme mit Sedimentationsplatten. Gemäß den UK GMP-Richtlinien sollten Sedimentationsplatten mindestens wöchentlich getestet werden, während nicht-lebensfähige Partikelzählungen täglich durchgeführt werden sollten. Die Überwachungshäufigkeit sollte nach Wartungsarbeiten erhöht werden [3][4]. Alle Daten sollten in Echtzeit protokolliert werden, mit definierten Alarmgrenzen. Beispielsweise könnte eine Zone der Klasse A ein Aktionslimit von 1 KBE/m³ für lebensfähige Partikel festlegen [1][2]. Die Analyse von Trends in diesen Daten kann helfen, potenzielle Probleme frühzeitig zu identifizieren.
Fortschrittliche Werkzeuge wie ferngesteuerte Laserpartikelzähler, aktive Luftprobenehmer und Datenlogger mit Echtzeitwarnungen gewährleisten eine kontinuierliche Überwachung.Drahtlose Sensornetzwerke bieten 24/7-Überwachung über Dashboards, wodurch die Abhängigkeit von manuellen Kontrollen reduziert wird [2][10]. Um die Genauigkeit zu gewährleisten, sollten Sensoren alle sechs Monate einer vorbeugenden Wartung unterzogen werden.
Neubewertungsplanung
Die Neubewertung stellt sicher, dass die Leistung des Reinraums innerhalb der erforderlichen Spezifikationen bleibt, selbst wenn Geräte altern, Prozesse sich entwickeln oder sich regulatorische Anforderungen ändern. Auslöser für die Neubewertung sind wesentliche Änderungen, wie die Installation neuer Bioreaktoren, die Aufrüstung von HLK-Systemen oder die Änderung von Anlagenlayouts. Für Anlagen zur Herstellung von kultiviertem Fleisch müssen auch Prozessänderungen - wie Modifikationen in Medienformulierung - berücksichtigt werden, um Kontaminationsrisiken zu managen [1] [3].
Kritische Parameter sollten jährlich neu validiert werden, mit halbjährlichen Überprüfungen und sofortiger Nevalidierung nach wesentlichen Änderungen. Gemäß den MHRA GMP-Richtlinien sollten Hochrisiko-Reinräume für kultiviertes Fleisch ihre Leistungsqualifikation (PQ) alle 12 Monate neu validieren, wobei alle IQ-, OQ- und PQ-Elemente abgedeckt werden. Nach HVAC-Verbesserungen sollte die erneute Prüfung innerhalb von 30 Tagen erfolgen [4] [10]. Präventive Wartungspläne sollten auch mit GMP-Audits übereinstimmen [2][3].
Für laufende Validierungsanforderungen verbindet
Compliance-Standards für Reinräume in der kultivierten Fleischproduktion
Nach der Behandlung von Validierungs- und Testprotokollen besteht die letzte Hürde für die Produktion von kultiviertem Fleisch darin, die Compliance-Standards zu erfüllen, um die behördliche Genehmigung zu sichern. Reinräume, die in diesem Prozess verwendet werden, müssen den ISO 14644 für Partikelgrenzen und Testmethoden entsprechen, zusammen mit den Good Manufacturing Practice (GMP) Richtlinien für Kontaminationskontrolle und Validierung. Durch die Einhaltung dieser Rahmenbedingungen können Hersteller sicherstellen, dass ihre Einrichtungen den strengen behördlichen Anforderungen entsprechen. Lassen Sie uns die Rolle jedes Standards in der Reinraum-Compliance aufschlüsseln.
ISO 14644 Standards für Reinraumklassifizierung
ISO 14644 beschreibt Reinraumklassifizierungen basierend auf der Konzentration von luftgetragenen Partikeln. Es misst Partikel mit einer Größe von ≥ 0,5 μm pro Kubikmeter, mit Klassen von ISO 1 (am saubersten) bis ISO 9. Für die Produktion von kultiviertem Fleisch sind die relevantesten Klassifizierungen ISO 5 bis ISO 8, die mit den GMP-Graden A bis D übereinstimmen. Diese Standards konzentrieren sich auf "Ruhezustände" - wenn der Reinraum vollständig eingerichtet, aber unbesetzt ist.
Während ISO 14644 die Grundlage für die Klassifizierung von Reinräumen bildet, deckt es keine Validierung während aktiver Operationen ab und erfordert keine mikrobiologische Überwachung. Hier kommen die GMP-Richtlinien ins Spiel, die eine zusätzliche Compliance-Ebene für Anlagen zur Produktion von kultiviertem Fleisch hinzufügen.
GMP-Anforderungen für kultiviertes Fleisch
Im Gegensatz zu ISO-Standards verlangt GMP die Validierung sowohl im "Ruhezustand" (unbesetzt) als auch im "Betrieb" (besetzt). Beispielsweise erlaubt ein Reinraum der Klasse B bis zu 3.520 Partikel ≥ 0,5 μm/m³ im Ruhezustand, aber diese Zahl erhöht sich auf 352.000 Partikel während des Betriebs [12] .
GMP verwendet eine Kontaminationskontrollstrategie (CCS), geleitet durch Qualitätsrisikomanagement (QRM), um Kontaminationsrisiken zu identifizieren und zu minimieren. Die Richtlinien spezifizieren auch strukturelle und Oberflächenanforderungen, um Partikelansammlungen zu verhindern und eine effektive Reinigung zu ermöglichen. Oberflächen müssen glatt, wasserdicht und langlebig sein, während Schiebetüren aufgrund von Reinigungsschwierigkeiten nicht empfohlen werden. Zusätzlich sind Waschbecken und Abflüsse in Bereichen der Klasse A und B verboten, um mikrobielle Reservoirs zu vermeiden.
Da Menschen für 75–80 % der während Reinrauminspektionen nachgewiesenen Partikel verantwortlich sind, [11], erzwingt GMP strenge Bekleidungsprotokolle und beschränkt den Zugang des Personals während kritischer Phasen der Leistungsqualifizierung (PQ).
Für Produkte, die eine sterile Handhabung erfordern, umfasst die GMP-Validierung aseptische Prozesssimulationen (Medienfüllungen), um zu bestätigen, dass der Produktionsprozess eine mikrobielle Kontamination verhindern kann. Die Umweltüberwachung ist ein weiterer kritischer Aspekt, der sowohl nicht-lebensfähige Partikel als auch lebensfähige Mikroorganismen abdeckt. Grade-A-Zonen erfordern eine kontinuierliche Überwachung, während Bereiche niedrigerer Grade häufig überprüft werden, um die Einhaltung der Vorschriften zu gewährleisten.
Verwendung von Cellbase für Reinraumvalidierungsressourcen
Die Beschaffung von Reinraumvalidierungsgeräten für Anlagen zur Herstellung von kultiviertem Fleisch kann ein schwieriger Prozess sein, hauptsächlich aufgrund der spezialisierten Überwachungswerkzeuge, die zur Erfüllung der ISO 14644- und GMP-Standards erforderlich sind. Allgemeine Laborausrüstungsplattformen führen diese Nischenartikel oft nicht, sodass Beschaffungsteams Lösungen aus fragmentierten Lieferantennetzwerken zusammenstellen müssen. Hier kommt
Zugang zu verifiziertem Equipment und Materialien
Nehmen Sie zum Beispiel ein Startup, das seine Validierungszeit erfolgreich verkürzt hat, indem es über
Vereinfachte Beschaffung für Validierungsbedarfe
Über die Bereitstellung verifizierter Ausrüstung hinaus macht
Einkaufsleiter haben eine schnellere Wiederauffüllung von wesentlichen Überwachungswerkzeugen gemeldet, einschließlich Echtzeit-Partikelzählern und Datenloggern, die entscheidend für die Aufrechterhaltung effektiver Umweltüberwachungsprogramme und die Planung der Revalidierung gemäß GMP-Richtlinien sind [18] . Zusätzlich unterstützt
Fazit
Die Reinraumvalidierung in der Produktion von kultiviertem Fleisch ist ein akribischer Prozess, der sicherstellt, dass die Einrichtungen die ISO 14644-Partikelgrenzen und GMP-Standards erfüllen, bevor Bioreaktoroperationen beginnen können. Die Daten sprechen für sich: Validierte Reinräume erreichen konsequent eine Sterilitätssicherungsrate von 99,99 %, wobei ISO 14644-konforme Einrichtungen Kontaminationsraten unter 1 % melden.Im Gegensatz dazu haben nicht validierte Umgebungen Kontaminationsraten von bis zu 15 % - ein deutlicher Unterschied, der die Bedeutung einer ordnungsgemäßen Validierung hervorhebt[13] [14].
Aber die Arbeit endet nicht nach der anfänglichen Validierung. Die Aufrechterhaltung der Reinraumleistung ist ebenso wichtig. Laut Experten des Cleanroom Technology Institute sind unzureichende Validierungen für 40 % der GMP-Nichtkonformitäten in der Biopharma verantwortlich. Für kultiviertes Fleisch stellt dies ein ernsthaftes Risiko dar, da bereits ein einziges Kontaminationsereignis Produktionsläufe im Wert von Zehntausenden von Pfund gefährden könnte, was die Notwendigkeit einer zuverlässigen Beschaffungsschicht zur Sicherung hochwertiger Eingaben[13][14].
FAQs
Was ist der Unterschied zwischen Qualifizierung und Validierung in einem Reinraum?
Qualifizierung und Validierung spielen unterschiedliche, aber gleichermaßen wichtige Rollen bei der Aufrechterhaltung der Reinraumkonformität.
Qualifizierung bezieht sich darauf, sicherzustellen, dass der Reinraum und seine Systeme ordnungsgemäß installiert sind und wie vorgesehen funktionieren. Dieser Prozess umfasst mehrere Phasen, einschließlich Designqualifizierung (DQ), Installationsqualifizierung (IQ), und Betriebsqualifizierung (OQ). Jeder Schritt bestätigt, dass der Reinraum seine Designvorgaben erfüllt und effektiv arbeitet.
Validierung, hingegen konzentriert sich auf die Fähigkeit des Reinraums, während der tatsächlichen Produktion kontinuierlich die erforderliche Umgebung bereitzustellen. Es geht darum, langfristige Zuverlässigkeit, Sicherheit und die Einhaltung von regulatorischen Standards sicherzustellen.
Wie wähle ich die richtige ISO-Klasse/GMP-Stufe für Bereiche der kultivierten Fleischproduktion aus?
Bei der Wahl der richtigen ISO-Klasse oder GMP-Stufe für die Produktion von kultiviertem Fleisch kommt es auf die spezifische Produktionsstufe und die damit verbundenen Kontaminationsrisiken an.
- ISO-Klasse 5: Am besten geeignet für frühe Kultivierungsphasen, in denen die Aufrechterhaltung der Sterilität entscheidend ist.
- ISO-Klasse 6: Ideal für Bioreaktoroperationen, die Sauberkeit mit Praktikabilität in Einklang bringen.
- ISO-Klasse 8: Geeignet für Ernte- und Transferprozesse, bei denen die Kontaminationsrisiken geringer sind.
Die Aufrechterhaltung höherer Sauberkeitsstandards ist in Bereichen, in denen die Sterilität nicht beeinträchtigt werden darf, unerlässlich. Darüber hinaus sind geeignete Umweltkontrollen erforderlich, um die behördlichen Anforderungen zu erfüllen.
Welche Änderungen erfordern eine sofortige Reinraum-Revalidierung?
Wenn wesentliche Änderungen vorgenommen werden - wie Änderungen am Reinraumlayout, die Hinzufügung neuer Geräte oder Aktualisierungen der Umweltkontrollen, die die Sterilität oder Compliance beeinflussen könnten - wird eine sofortige Revalidierung notwendig. Solche Änderungen können kritische Bedingungen beeinflussen, daher stellt die Revalidierung sicher, dass alles weiterhin den regulatorischen Anforderungen entspricht.










