

クリーンルームの認証は、培養肉の生産において重要であり、Regulation (EC) 853/2004のような英国の規制に対する安全性とコンプライアンスを保証します。認証がないと、施設は汚染、非コンプライアンス、製品品質の問題のリスクがあります。以下はプロセスの簡単な概要です:
- 認証が重要な理由: 微生物汚染を防ぎ、HACCP原則に沿い、一貫した生産を保証します。
- 主要な基準: ISO 14644-1(空気清浄度)、EU GMP Annex 1(無菌製造)、EN 17141(微生物管理)。
-
認証へのステップ:
- 設計と構築: HEPAフィルター、気流システムを設置し、適切な圧力差を維持します。
- 設置適格性評価 (IQ): クリーンルームが設計仕様に合致していることを確認します。
- 運用適格性評価 (OQ): 制御された条件下で性能をテストします。
- 性能適格性評価 (PQ): 実際の運用中の機能を検証します。
- 継続的モニタリング: 粒子、圧力、温度、湿度の定期的なチェック。
- 継続的なコンプライアンス: 6~12ヶ月ごと、または大きな変更後の再適格性評価。
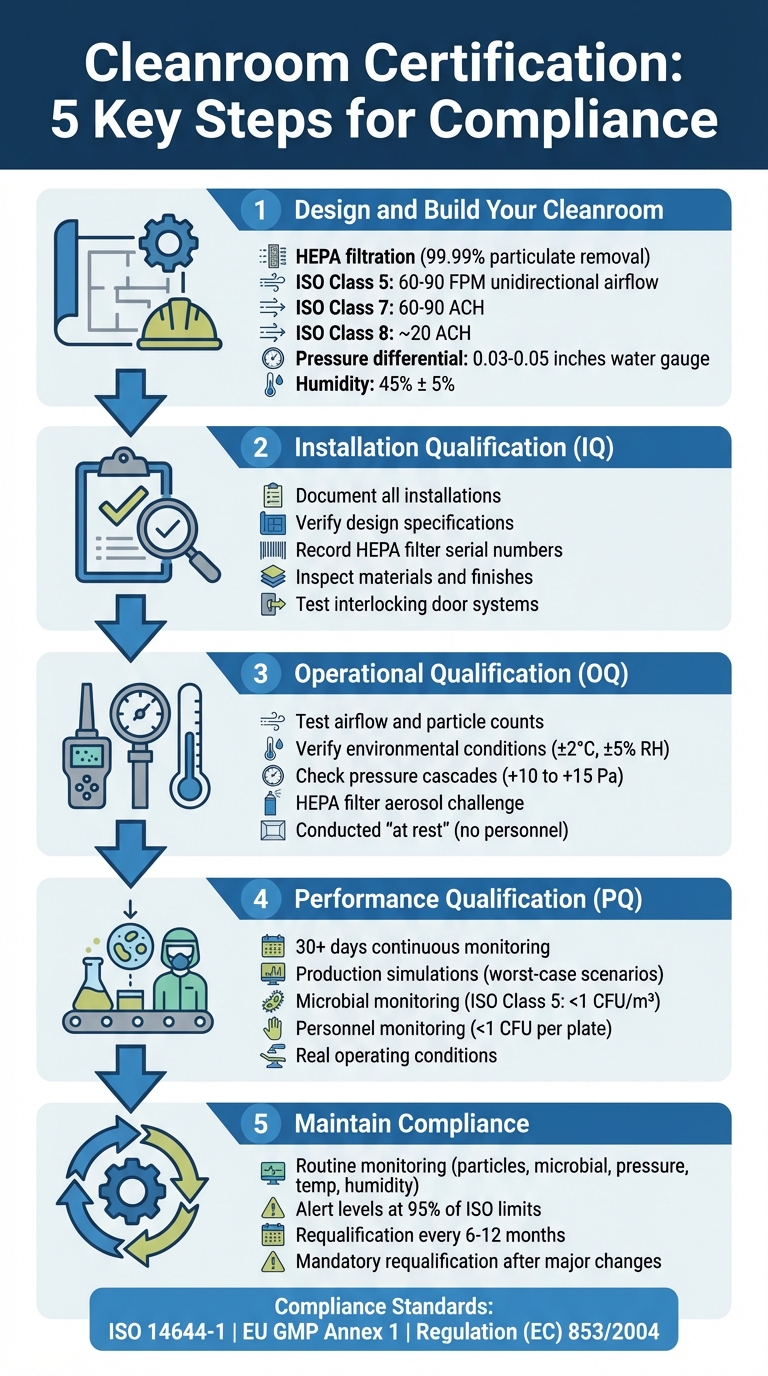
培養肉施設のための5ステップクリーンルーム認証プロセス
クリーンルームの設計と認証
ステップ1: クリーンルームの設計と構築
培養肉生産のためのクリーンルームを構築するには、HEPAフィルター、環境制御、および作業フローの分離の3つの主要システムに関する慎重な計画が必要です。これらの要素は無菌状態を確保し、後の高額な修正を避けるのに役立ちます。これらのシステムが導入されたら、設置中のパフォーマンスの最適化に注力してください。
HEPAフィルターと気流の要件
空気処理システムはクリーンルームの基盤であり、必要なISO分類を満たしているかどうかを決定します。HEPAフィルターは99.99%の微粒子を除去します[5]が、その効果は各生産ゾーンに合わせた特定の速度とパターンで空気を供給することに依存します。
ISOクラス5のエリアでは、肉の収穫のような無菌プロセスが行われるため、一方向(層流)気流を60から90フィート毎分(FPM) [5]で必要とします。これは、天井に取り付けられたファンフィルターユニット(FFU)が狭い放出角度で低壁の通気口を通して気流を指示することを含みます。
ISOクラス7および8のゾーンでは、混合気流設計が使用されます。ここでは、新鮮な供給空気が内部空気と混合され、1時間あたりの空気交換回数(ACH)が少なくて済みます。 ISOクラス7のゾーンは60〜90ACHが必要であり、ISOクラス8のゾーンは約20ACHが必要です[1][6]。ACHを計算するには、供給空気量(毎時)を部屋の体積で割ります。天井が高いと、気流の要求とコストが増加することに注意してください[3]。
圧力差は、汚染物質を排除するための鍵です。クリーンルームと隣接するエリアの間で0.03〜0.05インチ水柱の正圧差を維持してください [7]。ただし、ドアを越える圧力差が0.1インチ水柱を超えないようにしてください。これにより、標準的な3×7フィートのドアを開けるのに最大11ポンドの力が必要になる可能性があります[7]。
設置後、エアロゾルチャレンジテスト(DOPまたはPAOテストなど)を使用して、HEPAフィルターハウジングに漏れや弱いシールがないことを確認するために、ろ過システムの完全性をテストします[1]。煙の可視化研究は、層流の確認や、敏感なゾーンでの乱流や逆流がないことを確認するのにも役立ちます。
ろ過が整ったら、次のステップは一貫した環境条件を確保することです。
生産のための環境制御
温度、湿度、圧力を安定して維持することは、微生物リスクを最小限に抑えるために重要です[9]。HVACシステムは、クリーンルームのISO分類に必要な高い換気回数をサポートしながら、これらのパラメータを継続的に維持しなければなりません。
特に温度管理が重要です。システムは、バイオリアクター、照明、スタッフからの熱を相殺しながら、気流パターンを乱さないようにしなければなりません。相対湿度は45% ± 5%に保たれ、静電気による粒子の吸着を防ぎ、結露の問題を回避します。この範囲は、ガウンを着たスタッフにとって快適な作業環境を保証します[7].
pHや溶存酸素のような環境パラメータのリアルタイムモニタリングは、微生物の成長の初期兆候を検出するのに役立ちます[9]。空気、表面、水の継続的なモニタリングは、汚染が拡大する前に捕捉するためのもう一つの重要な実践です。さらに、HVACシステムを短い回復時間で設計してください - これにより、汚染イベント後にクリーンルームが迅速に指定された清浄度レベルに戻り、ダウンタイムを削減します[1].
環境管理が整ったら、材料や人の移動を管理し、汚染リスクを最小限に抑えることに集中してください。
材料と人員の流れの管理
"クリーンルームの作業者はクリーンルームの最大の汚染源であり、すべての重要なプロセスは人員アクセスドアや通路から隔離されるべきです。" - Vincent A. Sakraida, エンジニア[7]
人員はクリーンルームにおける最大の汚染リスクであり、皮膚の粒子、髪の毛、その他の破片を放出します[7][6]。これに対処するために、クリーンルームのレイアウトは重要なプロセスを高トラフィックエリアやアクセスポイントから物理的に分離する必要があります。
材料は一方向に移動するべきです - 低グレードゾーンから高グレードゾーンへ - その途中で検証された消毒または滅菌プロセスを使用して[8]。両端滅菌器、例えば両扉オートクレーブや除熱トンネルは、空気の質を損なうことなく無菌処理エリアに物品を移動させるのに理想的です[8].
エアロックは、異なる清浄度レベルのエリア間の緩衝として機能します。ISOクラス7またはそれ以上の清浄ゾーンには、外部の汚染物質が生産エリアに入るのを防ぐために、ガウン着用のための前室を含めるべきです[6]。グレードAおよびBゾーンでは、インターロックドアシステムにより、一度に一つのドアしか開けられないようにします[8]。人員と物品のための別々のエアロックが実現不可能な場合、手続き上の時間的分離により、ゾーン間の同時移動を避けることができます[8].
交差汚染のリスクを減らすために、重要なスペースへのアクセスを単一の出入口に制限します[7]。 観察窓やリモートカメラを使用することで、監督者はクリーンルームに入ることなく活動を監視でき、不要なアクセスを削減できます[8].
これらの各対策は、クリーンルームの認証を達成し、安全でコンプライアンスに準拠した培養肉の生産に必要な厳しい基準を満たすために重要な役割を果たします。
ステップ2: 設置適格性評価 (IQ) の完了
建設が完了したら、次のステップは設置適格性評価 (IQ) です。このプロセスは、クリーンルームのすべてのコンポーネントが正しく設置されていることを確認し、運用テストに進む前に行います。基本的に、IQは物理的な建設の完了とHVACのバランス調整の開始の間の橋渡しとして機能し、次の段階に進む準備が整っていることを確認します。
"設置適格性確認(IQ)チェックは、機器、コンポーネント、およびクリーンルームのセットアップがメーカーの仕様に準拠していること、そしてすべてが正しく設置されていることを確認します。" - Kjeld Lund, クリーンルームスペシャリスト [11]
IQは「完成時」の状態に焦点を当てています - クリーンルームの構造が完成しているが、生産設備がまだ設置されていない状態です。ここでの主な目的は、建設されたものが元の設計に一致していることを確認し、逸脱が適切に文書化され、対処されていることです。
すべての設置手順を文書化する
この段階では、徹底した文書化が重要です。詳細な完成時の記録が必要で、これには更新された建築図面、HVAC機器のリスト、制御シーケンス、および電気配線図が含まれます。これらの記録は、クリーンルームが実際にどのように建設されたかを反映している必要があり、計画された方法だけではありません。
各HEPAまたはULPAフィルターについて、シリアル番号、正確な場所、および設置日を記録してください。設置直後に輸送中の損傷がないかフィルターを検査することが重要です。小さな漏れでもクリーンルームの完全性を損なう可能性があります。すべての機器とセンサーに、機器リストと一致する一意のIDをタグ付けし、将来の監査とメンテナンスを簡素化します。
すべての監視機器の校正証明書もファイルに保管する必要があります。これには、パーティクルカウンター、差圧センサー、温度および湿度プローブ、気流装置が含まれます。ISOクリーンルームのトニ・ホースフィールドは、「パーティクルカウンターの校正証明書はクリーンルームのバリデーションレポートに含まれています」と説明しています。[10]
材料と仕上げの検査も同様に重要です。壁パネル、床、ドア、パススルー、シーラントがGMP基準を満たしていることを確認してください。表面は、非脱落性、低VOC、適切にシールされている必要があります。ドアと窓は、圧力の整合性を維持するために壁と面一でなければなりません。
設計のばらつきに関する逸脱ログを保持し、評価と取られた是正措置を記録します。このログは、インストールフェーズからのすべての発見を統合する最終的な検証レポートの一部となります。
設計仕様を確認する
インストール記録が完了したら、次のステップはすべてのシステムが承認された設計と一致していることを確認することです。ユーザー要求仕様書(URS)と物理的なインストールを照合し、出荷や組み立て中に見落としがないことを確認します。
HVACおよびろ過システムについては、空気処理ユニット、ダクト接続、およびディフューザーの位置が設計図と一致していることを確認します。HEPAフィルターがハウジングに正しく設置されていること、およびすべてのダクトワーク圧力テストが正常に完了していることを確認してください。各ファンフィルターユニットの仕様とデータシートを記録します。
構造の検証には、インターロック、エアロック、およびパススルーが意図したとおりに動作することを確認するための検査が含まれます。インターロックドアシステムをテストして、両方のドアが同時に開かないことを確認します。すべてのシールが無傷であり、クリーンルームが必要な圧力差を維持できることを確認してください。
次のテスト段階に進む前に、HVACシステムを運転して定常状態を達成します。
精密にIQを実施することは重要であり、それがすべての後続の資格段階の基礎を築きます。ステップをスキップしたり、文書化を急いだりすると、運用テストや規制監査中に問題が発生する可能性があります。これらのチェックを徹底的に完了することで、運用資格へのスムーズな移行を確保します。
ステップ3: 運用適格性評価 (OQ) を実施する
設置適格性評価がすべてが正しく設置されていることを確認したら、次のステップは運用適格性評価 (OQ) です。このフェーズでは、定義された条件下でクリーンルームが意図した通りに動作することを確認します。通常、これらのテストは「静止状態」で行われ、HVACシステムは稼働していますが、スタッフや生産活動は行われていません。
「バリデーションは、クリーンルームが設計された通りに機能し、静的(静止状態)および運用条件の両方で安定した、汚染物質のない環境を維持することを客観的に証明します。」 - Standard Tech [12]
OQテストは、ISO 14644-1およびGMP基準への準拠を示すために重要です。培養肉施設にとって、このステップは特に重要であり、生物学的プロセスは厳格な粒子および微生物制御に依存しています。正確な結果を確保するために、テストを開始する前にクリーンルームを少なくとも30分間安定させ、粒子数の偏りを避けてください[12]。これらのチェックは設置段階に基づいており、環境制御の微調整の基礎を築きます。
気流と粒子数のテスト
空中粒子のカウントはISO分類の基礎です。校正されたレーザー粒子カウンターを使用して、空気中の粒子濃度を測定し、必要なISOクラスへの準拠を確認します。ISOクラス5の場合、基準表に記載されている粒子限界を参照してください。
サンプリング場所の数はクリーンルームのサイズに依存します。ISO 14644-1は明確なガイドラインを提供しており、大きな部屋にはグリッドパターンでより多くのサンプリングポイントが必要です[16]。2から9か所をテストする場合、準拠を判断するために95%上限信頼限界(UCL)を計算する必要があります。10個以上のサンプリングポイントがある場合、この計算は不要です[15].
気流速度と体積の測定は、空気交換率が設計仕様を満たしていることを確認します。アネモメーターを使用して、特に重要なプロセスエリア付近のさまざまなポイントで速度を測定し、これらの値が設計目標と一致していることを確認してください。
煙の研究は、気流の方向を確認する視覚的な方法を提供し、より清潔なゾーンからあまり清潔でないゾーンへと移動することを保証します。ドア、パススルー、およびその他の脆弱なエリアの近くで煙を発生させ、気流を乱す可能性のある漏れや乱流を検出します[12]。粒子カウンターは正確ですが、煙の研究は、他の方法では見逃される可能性のある停滞ゾーンのような問題を明らかにすることができます。
HEPAおよびULPAフィルターもOQ中に再検証する必要があります。エアロゾルチャレンジを使用して、フィルターまたはそのシールに漏れがないか確認します。設置のわずかな誤りでも性能に影響を与える可能性があるため、メンテナンスやフィルター交換後には必ず再テストを行ってください [12].
気流性能が確認されたら、製品の品質と作業者の快適さに影響を与える環境条件に焦点を移します。
環境条件の確認
温度と湿度は、製品の品質を維持し、快適な作業環境を確保する上で重要な役割を果たします。培養肉施設では、温度の検証目標は通常±2°C、相対湿度は±5%です [12]。スポットチェックでは見逃される可能性のある変動を検証するために、少なくとも24時間の継続的な監視が推奨されます [12].
培養肉用クリーンルームは通常、温度範囲18–22°C、相対湿度30–60%を維持します [14]。これらの条件は、微生物の成長を促進する可能性のある結露を防ぎながら、細胞培養プロセスをサポートします。クリーンルーム全体に配置された校正済みの温度センサーとRHプローブを使用して、条件の変動を特定します。
圧力カスケードは、もう一つの重要な要素です。これにより、空気がより清潔なエリアから清潔度の低いエリアへと流れ、汚染リスクが低減されます。隣接する分類された部屋間で一般的に+10から+15 Paの圧力差を校正済みのゲージを使用して確認します。ドアやパススルーでの測定を定常状態で行い、適切な圧力関係を確認します[12].
回復時間テストは、汚染イベント後にクリーンルームがどれだけ早く基準に戻るかを測定します。制御された粒子源を導入し、粒子数が基準値に戻るまでの時間を監視します。回復時間が速いことは、より良い気流設計とより効果的な汚染制御を示しています[1].
遅延や高額な再試験を避けるために、OQテストの直前にすべての機器を校正してください。日付、時間、場所、機器ID、各テストの環境条件を含むすべての関連詳細を記録します。この文書は、検証レポートに不可欠であり、規制監査の際に必要となります[12].
sbb-itb-ffee270
ステップ4: パフォーマンス適格性評価 (PQ) を実施する
パフォーマンス適格性評価 (PQ) は、実際の生産条件下で、機器が稼働し、スタッフが積極的に作業している状態でクリーンルームの性能を評価します[1][12].インストールおよび運用資格に基づいて、PQはクリーンルームが実際の運用中に一貫して信頼性のあるパフォーマンスを提供することを確認します。
"PQは、機器の操作や人員の活動を含む実際の運用条件下でのクリーンルームの性能を検証します。" - G-CON [1]
徹底的なテストを確保するために、PQフェーズには少なくとも30日間の連続監視を含めるべきです。この延長された期間は、短期間のテストでは見落とされる可能性のある、製造サイクル中の温度変動やスタッフの動きによる微生物汚染の変化などの変動を特定するのに役立ちます。厳格な汚染管理が重要な培養肉施設において、PQはクリーンルームが日常の運用中に準拠していることを文書で証明します。
生産シミュレーションを実行する
生産シミュレーションは最悪のシナリオを再現する必要があります。これには、最大収容人数、すべての機器の同時運転、頻繁なドアの開閉や激しい動きなどの一時的な汚染リスクが含まれる可能性があります[1][13]。故障モード影響解析(FMEA)などのリスクベースのアプローチは、汚染リスク、物質の流れ、交通量の多いエリアに基づいてサンプリング場所を特定するのに役立ちます[16]。
これらのシミュレーション中の微生物モニタリングは重要です。コロニー形成単位(CFU)は、アクティブおよびパッシブのサンプリング方法の両方を使用して追跡する必要があります[14][17]。ISOクラス5のクリーンルームでは、微生物汚染のアクションリミットは一般的に1 CFU/m³です[14]。
クリーンルームでの最大の粒子源は人間であるため、個人のモニタリングも同様に重要です。手袋をした指先のサンプリングは、適切な無菌技術を確認することができ、プレートあたり1 CFU未満が許容限界です[17]。オペレーターは、汚染レベルの人工的なスパイクを防ぐために、事前にガウンの着用と動作プロトコルについて説明を受けるべきです[12]。
さらに、制御された粒子チャレンジ後のクリーンルームの回復速度をテストします。粒子源を導入し、条件が基準に戻るまでの時間を測定します。このプロセスは、気流と汚染制御システムの効果を評価します[1][12]。
環境モニタリングの設定
シミュレーションテストの後、継続的な環境モニタリングは一貫したパフォーマンスを保証します。これらのシステムは、浮遊粒子、微生物汚染、温度、湿度、圧力差などの重要なパラメータに関するリアルタイムデータを提供します。これは、コンプライアンス問題に至る前にパフォーマンスの変化を検出するために不可欠です[1]。培養肉の生産において、継続的なモニタリングは不可欠です。
資格認定フェーズでは、効果的な汚染管理を確認するために、重要なゾーンで1~2時間ごとに微生物の空気サンプリングを実施します[14]。細菌を検出するためにトリプチックソイ寒天(TSA)を使用し、サンプルを30~35°Cで最低3日間培養し、真菌やカビにはサブロー寒天(SAB)を使用し、20~25°Cで少なくとも7日間培養します[17]。テスト中は、エアサンプラーの近くでエアロゾルスプレーや消毒剤の使用を避けてください。回転式消毒剤または70%イソプロピルアルコールが適用された場合、空気サンプリングを開始する前に少なくとも5分待ってください[17].
監視されるすべてのパラメータに対して明確な警告および行動限界を設定します。警告レベルは、値が逸脱し始めたときに調査の必要性を示し、行動限界は、パラメータが許容範囲を超えた場合に即時の是正措置を要求します[14]。日付、時間、場所、機器の詳細、環境条件を含むすべての測定の詳細な記録を保持します。これにより、常に監査に備え、ISO 14644およびGMP基準への準拠を示すことができます。
ステップ5: 監視を通じたコンプライアンスの維持
性能適格性評価を完了したら、そこで作業が終わるわけではありません。コンプライアンスを維持するには継続的な監視と定期的な再評価が必要です。クリーンルームの認証は一度きりのマイルストーンではなく、施設を「管理状態」に保つために一貫した努力が求められます。培養肉施設において、この継続的なプロセスは、規制および運用基準の両方を満たし、資格取得段階の綿密な実践を日常業務に拡張することを保証します。
定期的なモニタリングの実施
クリーンルームがISO 14644およびGMP基準に準拠し続けるためには、微生物および粒子状汚染物質を定期的に監視する必要があります。注視すべき主要なパラメータには以下が含まれます:
- 粒子数
- 微生物レベル
- 圧力
- 温度
- 湿度
- 気流
モニタリングの頻度は、クリーンルームの分類と徹底的なリスク評価に合わせる必要があります。例えば、ISOクラス5のゾーンでは、製造中に連続または毎時の粒子モニタリングが必要なことが多いですが、重要度の低いエリアでは、毎日または毎週のチェックのみが必要な場合があります。
ISOの限界値の95%で警報レベルを設定し、潜在的な問題を早期にキャッチします。これらのレベルは、パラメータがずれ始めたときに警告として機能し、問題が拡大する前に調査を促します。一方で、アクションリミットは、パラメータが許容範囲を超えた場合に即時の是正措置を要求します [14] 。
モニタリングのもう一つの重要な部分は、手袋をした指先のサンプリング(GFS)です。この方法は、スタッフが適切な無菌技術を維持していることを確認します。標準基準は通常、プレートあたり1 CFU未満です[17]。重要な無菌作業の後や各シフトの終わりにGFSを実施することで、技術の欠陥を早期に特定し対処することができます。
日常的なモニタリングは日々の管理を維持するのに役立ちますが、再認定はクリーンルームシステムが長期にわたって効果的であることを保証します。
再認定のスケジュールを立てる
再認定は6〜12ヶ月ごとに実施する必要があります。ただし、構造の改修、新しい機器の設置、HEPAフィルターの交換、またはHVACシステムの大幅な変更など、特定のイベントが発生した場合は再認定が必須です[1][14]。
再認定中には、運用資格フェーズの多くのテストを再実施する必要があります。これには以下が含まれます:
- 空中粒子のカウント
- HEPAフィルターの完全性試験(粒子≥0に対して99.99%の効率を証明する)。3 microns)
- 気流速度測定
- 圧力差チェック
特に重要なテストの一つは、回復時間テストであり、汚染イベント後にクリーンルームが目標の清浄度レベルにどれだけ早く戻るかを測定します。このテストは、HVACシステムがストレスを効果的に処理する能力を検証します[1].
バリデーションマスタープラン (VMP)を保持し、すべての適格性確認段階(IQ、OQ、PQ)と再適格性確認スケジュールを文書化します。テストに使用するすべての機器 - パーティクルカウンターや風速計など - が校正され、国家標準に追跡可能な証明書を持っていることを確認してください。これにより、コンプライアンスの取り組みにおける正確性と信頼性が保証されます[1] [14].
コンプライアンスのためのクリーンルーム機器を調達
認定されたサプライヤーを見つける Cellbase
クリーンルームの運用および性能基準が確立され、検証されたら、次のステップはコンプライアンスを維持するための適切な機器を調達することです。培養肉の生産においては、業界の特有の要求を理解しているサプライヤーと協力することを意味します。
GMP準拠の調達を確保する
機器を確認した後、調達プロセスも厳格なGMP基準を満たす必要があります。
さらに、プラットフォームはGMP要件を満たす材料を優先します。例えば、316Lステンレス鋼のような建設材料は、研磨された非脱落性の表面を持ち、繰り返しの化学消毒に耐性があり、検証済みの定置洗浄(CIP)および定置滅菌(SIP)プロトコルをサポートします [4]。材料の適合性を最初に確認することで、後で高価な改造や再評価サイクルを避けることができます。この積極的なアプローチは、コンプライアンスを維持しながら、時間とリソースを節約するのに役立ちます。
結論
重要なポイント
クリーンルームの認証を取得することは、製品の品質を維持し、規制基準を満たすために重要です。これは、HEPAフィルター、制御された気流、効率的な材料の流れを備えたクリーンルームの設計から始まります。このプロセスは、設置適格性評価 (IQ)、運用適格性評価 (OQ)、性能適格性評価 (PQ)の3段階の適格性評価アプローチで続きます。これらの段階は、すべてのシステムが実際の作業条件下で効果的に動作することを保証します。
認証はそこで終わりません。温度、湿度、圧力、粒子数などの要因の継続的な監視は、性能の問題を特定するために不可欠です。定期的な再検証は、ISO 14644-1およびGMP基準への準拠を保証し、将来の改善のための堅固な枠組みを作成します。
施設の次のステップ
これらの基準に施設を合わせるために、バリデーションマスタープラン (VMP)の開発を検討してください。このプランは、資格認定プロセスを日常の運用ニーズと統合し、規制の要求に先んじるのに役立ちます[1]。さらに、HACCPベースの食品安全管理システムの導入が重要です。少なくとも1人のチームメンバーがHACCP原則のレベル4に訓練され、コンプライアンスを確保する必要があります[2]。
設備のニーズについては、
よくある質問
培養肉生産におけるクリーンルーム認証の利点は何ですか?
クリーンルーム認証は、厳格な安全性と環境基準を遵守することで、培養肉生産において重要な役割を果たします。認証されたクリーンルームは、微生物や粒子からの汚染リスクを最小限に抑えるように設計されており、細胞培養に不可欠な無菌状態を維持します。これにより、最終製品の品質と安全性が保護されるだけでなく、ISO分類やGMPグレードなどの国際的に認められた基準を遵守し、規制当局の承認と市場での受け入れを確保します。
遵守を超えて、認証は、気流、ろ過、環境モニタリングなどの重要なシステムを検証することで、運用の信頼性を向上させます。これらのシステムは連携して汚染リスクを低減し、一貫した生産を可能にし、全体的なプロセス効率を向上させます。認定されたクリーンルームは、利害関係者に自信を与え、規制検査を簡素化し、管理された環境の管理におけるベストプラクティスの遵守を示すことで、スケーリングの取り組みをサポートします。
クリーンルームはどのくらいの頻度で再認定されるべきですか?
クリーンルームは、業界標準を満たしていることを確認するために定期的に再認定される必要があります。この頻度は、クリーンルームの分類、使用方法、リスク評価や環境モニタリング計画の結果など、いくつかの要因に依存します。
通常、再認定は年に一度行われます。しかし、高リスクの環境や、設備のアップグレードやレイアウトの調整などの大きな変更がある場合は、より頻繁なチェックが必要になることがあります。気流、ろ過、環境制御が必要な基準を引き続き満たしていることを確認するために、継続的な性能モニタリングも重要です。
培養肉生産のクリーンルームで監視すべき環境要因は何ですか?
培養肉施設でのコンプライアンスを確保し、汚染リスクを低減するためには、いくつかの環境要因を注意深く監視することが重要です。これには、粒子数、微生物汚染、気流パターン、気圧差、温度、および湿度レベルが含まれます。これらの要素を定期的に監視することで、GMP基準を維持し、生産に不可欠な制御された環境を作り出すことができます。
これらの条件を慎重に管理することで、施設はクリーンルーム認証に必要な厳しい基準を満たしながら、製品の品質を保護することができます。









