

クリーンルームのバリデーションは、培養肉の生産において厳しい汚染基準を満たすために、生産環境が重要であることを保証します。これは、培養肉のプロセスを拡大する. 際の重要なステップです。適切なバリデーションは、汚染リスク, を防ぎ、製品の品質を保護し、ISO 14644やGMPのような規制に準拠します。このプロセスは、次の4つの主要なフェーズで構成されます:
- 設計適格性評価 (DQ): クリーンルームの設計が運用および規制のニーズを満たしていることを確認します。
- 設置適格性評価 (IQ): コンポーネントが正しく設置され、仕様に一致していることを検証します。
- 運用適格性評価 (OQ): システムが意図した通りに機能することを確認するために、非稼働状態でテストします。
- 性能適格性評価 (PQ): 実際の生産中にクリーンルームの性能を評価します。
粒子数、HEPAフィルターの整合性チェック、気流測定を含む試験プロトコルは、コンプライアンスを維持するために重要です。継続的な監視と定期的な再検証は、クリーンルームの性能を長期間にわたって維持するのに役立ちます。これらの手順を遵守することで、汚染リスクが最小限に抑えられ、製品の一貫性と規制承認が保護されます。
URSからPQまでのクリーンルームバリデーション
sbb-itb-ffee270
クリーンルームバリデーションの4つのフェーズ
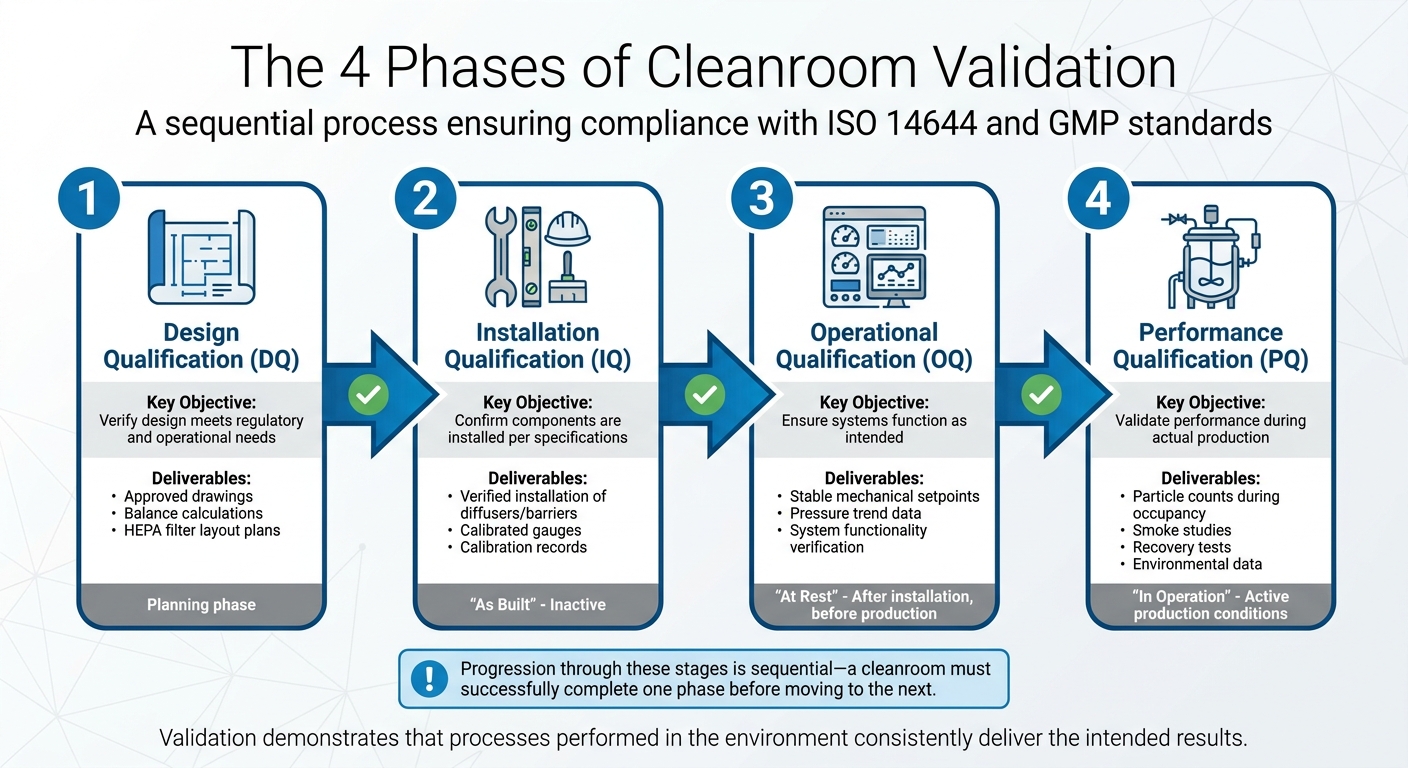
培養肉生産のためのクリーンルームバリデーションの4つのフェーズ
クリーンルームバリデーションは、段階的なプロセスであり、4つの異なるフェーズで構成され、それぞれが前のフェーズに基づいています。これらの段階は順次進行し、クリーンルームは次のフェーズに進む前に1つのフェーズを成功裏に完了しなければなりません。Allied Cleanroomsが適切に述べているように:
"バリデーションは、見た目が準備ができているクリーンルームと、実際に準備ができているクリーンルームを区別するものです"[8].
適格性評価は、クリーンルームとそのシステムが設計通りに設置され、機能することを保証しますが、バリデーションはさらに一歩進んでいます。それは、環境内で行われるプロセスが一貫して意図した結果をもたらすことを示しています[7]. 4つのステージ - 設計適格性評価 (DQ)、設置適格性評価 (IQ)、運用適格性評価 (OQ)、性能適格性評価 (PQ) - は、バリデーションされた生産プロセスのために施設を準備するように設計されています。これらのステージはまた、厳格なテストプロトコルの基礎を築きます。
| 検証フェーズ | 主要目的 | 一般的な成果物/テスト |
|---|---|---|
| 設計適格性評価 (DQ) | 設計が規制および運用のニーズを満たしていることを確認します。 | 承認された図面、バランス計算、HEPAフィルターレイアウト計画。 |
| 設置適格性評価 (IQ) | コンポーネントが仕様に従って設置されていることを確認します。 | ディフューザー/バリアの設置確認、校正済みゲージ。 |
| 運用適格性評価 (OQ) | システムが意図した通りに機能することを確認します。 | 安定した機械的設定値、圧力トレンドデータ。 |
| 性能適格性評価 (PQ) | 生産/占有中の性能を検証します。 | 粒子数、煙の研究、回復試験、環境モニタリング データ。 |
設計適格性評価 (DQ)
設計適格性評価 (DQ) フェーズは、クリーンルームの設計が培養肉生産の特定の要件に合致していることを確認します。これには、バランス計算やHEPAフィルタのレイアウトなどの設計文書が実際の運用ニーズを反映していることを検証することが含まれます。各設計要素は、ISO 14644基準またはユーザー定義の要件に基づく厳格な受け入れ基準を満たす必要があります [7].
設置適格性評価 (IQ)
設置適格性評価 (IQ) は、クリーンルームの「完成時」状態を非稼働状態で検証することに焦点を当てています。このフェーズでは、ディフューザー、リターン、およびバリアが設計仕様に一致していることを確認します。また、圧力モニターとゲージが正しく校正され、完全に動作していることも確認します。詳細な文書化、校正記録およびテスト場所のマッピングは、この段階において重要です [7][8].
運用適格性評価 (OQ)
運用適格性評価 (OQ) は、クリーンルームが「静止」状態にあるとき、つまり設置後で生産が始まる前にテストを行います。この段階では、システムが意図した通りに機能していることを確認するために、安定した機械的設定値と一貫した圧力傾向を文書化します。機器の移動や気流の変更など、重要な変更が発生した場合は、バランスを維持するためにターゲットを絞った再テストが必要です [7][8]. システムが正しく動作することが確認されると、施設は稼働条件下での性能検証の準備が整います。
性能適格性評価 (PQ)
最終段階である性能適格性評価 (PQ) は、実際の生産条件下でのクリーンルームの性能を検証します。このフェーズでは、培養肉生産のために使用されている施設がパフォーマンス目標を満たしているかどうかを評価します。主な評価には、占有中の粒子数、重要なエリア周辺の気流の可視化(煙の研究など)、および乱れの後に部屋が必要な清潔さに戻るまでの回復テストが含まれます。PQを開始する前に、機械的な設定値が安定していることを確認し、バイオプロセス制御ソフトウェア, を介して管理され、重要なサンプリング場所が特定され、清掃記録が検証された条件を確認します[7].
培養肉施設では、独立した第三者の検証機関を使用することが強く推奨されます。この公平な検証は、規制当局や監査人にとってより重みがあります。Allied Cleanroomsは次のように強調しています:
"規制当局や監査人は、結果に利害関係のない外部の当事者からの結果により重みを置きます" [8].
この独立したアプローチは、USDAの検査許可を求める施設にとって特に重要です。これは、FDAの事前市場相談を成功裏に完了することを要求します。 [5] [6].
クリーンルーム検証のための必要な試験プロトコル
設計適格性評価(DQ)、設置適格性評価(IQ)、運用適格性評価(OQ)、性能適格性評価(PQ)が完了したら、次のステップはクリーンルームの性能を確認するための一連の徹底的な試験です。これらの試験は、クリーンルームがそのISO分類に準拠し、培養肉の生産に適していることを保証します。以下に、主要な試験プロトコルの概要を示します。
浮遊粒子数試験
この試験は、クリーンルームがそのISO分類に準拠していることを確認するために、空気中の粒子の数を測定します。例えば、ISO 5クリーンルームは、1立方メートルあたり0.5 µm以上の粒子が3,520個を超えてはなりません。テストは、指定されたサンプリングポイントで校正された粒子カウンターを使用し、「静止状態」と「稼働状態」の両方で行います。ISO 14644-2によれば、ISO 5およびそれより厳しい分類では6ヶ月ごとに、ISO 6以上では年に一度、粒子濃度テストを実施する必要があります。[8].
HEPAフィルターの完全性テスト
これらのテストは、高効率微粒子空気(HEPA)フィルターが適切に機能しており、漏れや欠陥がないことを確認します。粒子数テストが部屋全体の清浄度を評価するのに対し、完全性テストはフィルター自体に焦点を当てます。フィルターの交換や部屋の改造などの重要な変更があった場合は、直ちに再テストが必要です。多くの施設は、これらのテストを実施するために第三者機関を選択します。独立した検証は、規制当局によって高く評価されることが多いためです。[8].
気流速度と体積の測定
適切な気流は清潔さを維持するために重要です。単方向のクリーンルームの気流は通常、0.45 m/s ±20%(0.36から0.54 m/sの間)であるべきです。測定は通常、作業高さで行われます。これは、スケーラブルなバイオリアクターシステム内でのバイオリアクター接種などの敏感な操作が行われる場所です。または、フィルター面から150〜300 mmの位置で行われます。ISO 14644-3:2005は、サンプリングポイントの数は部屋の面積(平方メートル)に10を掛けた平方根に等しく、最低4回の測定とフィルターごとに少なくとも1つのポイントが必要であると規定しています。煙の研究や気流の可視化マッピングは、一方向の気流をさらに検証し、「ウェイク領域」として知られる停滞した空気の領域を検出することができます。[9] .
圧力差のチェック
クリーンルームゾーン間の適切な圧力差を維持することは、汚染を防ぐために不可欠です。より清潔なゾーンは、隣接する清潔度の低いエリアに対して正圧を維持する必要があります。校正された圧力計とセンサーが使用され、安定した圧力差を文書化し、確保します。
温度と湿度の検証
クリーンルームの温度と湿度レベルは、培養肉の生産をサポートするために慎重に管理する必要があります。これらの条件は、製品の品質やHEPAフィルターおよびその他のシステムの性能に影響を与えます。継続的な監視により、これらのパラメータが生産サイクル全体で必要な設定値内に留まることを保証します。
継続的な監視と再検証
システムが導入された後も、検証は終わりません。フィルターの摩耗、HVACシステムの劣化、プロセスの変更の影響に対抗するためには、継続的な監視と定期的な再検証が不可欠です。DQ、IQ、OQ、PQを通じて初期のコンプライアンスを達成した後、アクティブな生産中のパフォーマンスを維持するには、継続的な監視が必要です。
環境モニタリングプログラム
堅牢な環境モニタリングプログラムは、定義されたスケジュールに従って、空中粒子数、微生物汚染、温度、湿度、圧力差を追跡します。グレードAゾーンでは、監視は継続的でなければならず、グレードBゾーンでは15〜30分ごとにチェックが必要です。グレードCおよびDゾーンは、リスク評価に基づいて、毎時またはシフトごとに監視することができます[3][4].
微生物モニタリングは、アクティブエアサンプリングとセトルプレートを組み合わせたものです。英国GMPガイドラインによれば、セトルプレートは少なくとも週に一度テストされるべきであり、非生存粒子のカウントは毎日行われるべきです。保守活動後にはモニタリングの頻度を増やす必要があります[3][4]. すべてのデータはリアルタイムで記録され、定義されたアラート限界を持つべきです。例えば、グレードAゾーンでは、生存粒子のアクション限界を1 CFU/m³に設定することがあります[1][2]. このデータのトレンドを分析することで、潜在的な問題を早期に特定するのに役立ちます。
リモートレーザーパーティクルカウンター、アクティブエアサンプラー、リアルタイムアラート付きデータロガーのような高度なツールは、継続的なモニタリングを保証します。ワイヤレスセンサーネットワークは、ダッシュボードを通じて24時間365日の監視を提供し、手動チェックへの依存を減らします[2][10]. 正確性を維持するために、センサーは6ヶ月ごとに予防保守を受けるべきです。
再検証スケジューリング
再検証は、設備が古くなったり、プロセスが進化したり、規制要件が変わったりしても、クリーンルームの性能が必要な仕様内に収まることを保証します。再検証のトリガーには、新しいバイオリアクターの設置、HVACシステムのアップグレード、施設レイアウトの変更などの大きな変更が含まれます。培養肉施設では、メディアの配合の変更などのプロセス変更も、汚染リスクを管理するために考慮する必要があります[1] [3].
重要なパラメータは毎年再検証されるべきであり、半年ごとのチェックと重要な変更後の即時再検証が必要です。MHRA GMPガイドラインによれば、培養肉の高リスククリーンルームは、すべてのIQ、OQ、およびPQ要素をカバーするパフォーマンス適格性評価(PQ)を12か月ごとに再検証する必要があります。HVACのアップグレード後、30日以内に再テストを行うべきです[4] [10]. 予防保全スケジュールもGMP監査に合わせるべきです[2][3].
継続的なバリデーションのニーズに対して、
培養肉クリーンルームのコンプライアンス基準
検証およびテストプロトコルに対処した後、培養肉生産の最終的なハードルは、規制当局の承認を得るためのコンプライアンス基準を満たすことです。このプロセスで使用されるクリーンルームは、粒子限界と試験方法に関するISO 14644に準拠し、汚染管理と検証のためのGood Manufacturing Practice (GMP) ガイドラインに従わなければなりません。これらのフレームワークに従うことで、製造業者は施設が厳しい規制要求を満たしていることを確認できます。それでは、クリーンルームコンプライアンスにおける各基準の役割を分解してみましょう。
ISO 14644 クリーンルーム分類の基準
ISO 14644は、空中浮遊粒子の濃度に基づいてクリーンルームの分類を示しています。0.5 μm以上の粒子を立方メートルあたりで測定し、クラスはISO 1(最も清浄)からISO 9まであります。培養肉の生産において最も関連性のある分類は、GMPグレードAからDに対応するISO 5からISO 8です。これらの基準は、クリーンルームが完全にセットアップされているが無人の「静止」状態に焦点を当てています。
ISO 14644はクリーンルームの分類の基礎を確立しますが、稼働中の検証や微生物モニタリングを要求するものではありません。ここでGMPガイドラインが役立ち、培養肉施設に対する追加のコンプライアンス層を提供します。
培養肉のGMP要件
ISO規格とは異なり、GMPは「静止状態」(無人)と「稼働状態」(有人)の両方の検証を義務付けています。例えば、グレードBのクリーンルームでは、静止状態で0.5μm/m³以上の粒子が最大3,520個許可されますが、稼働状態では352,000個に増加します[12] .
GMPは品質リスク管理(QRM), に基づく汚染制御戦略(CCS)を採用し、汚染リスクを特定し最小化します。ガイドラインはまた、粒子の蓄積を防ぎ、効果的な清掃を可能にするための構造および表面の要件を指定しています。表面は滑らかで防水性があり耐久性がある必要があり、清掃が困難なため引き戸は推奨されません。さらに、微生物の貯留を避けるために、グレードAおよびBのエリアではシンクや排水口は禁止されています。
クリーンルーム検査中に検出される粒子の75〜80%は人間が原因であるため、[11], GMPは厳格なガウン着用プロトコルを施行し、重要なパフォーマンス適格性評価(PQ)フェーズ中の人員アクセスを制限します。
無菌処理が必要な製品の場合、GMP検証には、無菌プロセスシミュレーション(メディアフィル)を含み、生産プロセスが微生物汚染を防ぐことができることを確認します。環境モニタリングはもう一つの重要な側面であり、非生存粒子と生存微生物の両方をカバーします。グレードAゾーンは継続的なモニタリングが必要であり、低グレードエリアはコンプライアンスを維持するために頻繁なチェックを受けます。
クリーンルーム検証リソースのためのCellbase
培養肉施設のためのクリーンルーム検証機器の調達は、ISO 14644およびGMP基準を満たすために必要な専門的な監視ツールのため、難しいプロセスとなることがあります。一般的なラボ供給プラットフォームは、これらのニッチなアイテムを在庫に持たないことが多く、調達チームは断片的なサプライヤーネットワークからソリューションを組み立てる必要があります。ここで登場するのが、培養肉産業に特化した初のB2Bマーケットプレイスである
検証済みの機器と材料へのアクセス
例えば、
バリデーションニーズのための簡素化された調達
検証済みの機器を提供するだけでなく、
調達マネージャーは、GMPガイドラインに基づく効果的な環境モニタリングプログラムの維持と再検証のスケジュール設定に重要なリアルタイム粒子カウンターやデータロガーを含む必須モニタリングツールの再入荷が迅速化されたと報告しています。 [18] . さらに、
結論
培養肉生産におけるクリーンルームのバリデーションは、バイオリアクターの運用を開始する前に施設がISO 14644の粒子限界とGMP基準を満たすことを確認するために設計された綿密なプロセスです。データが示す通り、バリデーションされたクリーンルームは一貫して99.99%の無菌保証率を達成しており、ISO 14644に準拠した施設は1%未満の汚染率を報告しています。対照的に、非検証環境では汚染率が最大15%に達することがあり、適切な検証の重要性を浮き彫りにしています[13] [14].
しかし、初期検証後の作業は終わりではありません。クリーンルームの性能を維持することも同様に重要です。クリーンルーム技術研究所の専門家によると、不十分な検証はバイオ医薬品におけるGMP不適合の40%を占めています。培養肉にとって、これは深刻なリスクをもたらします。たった一度の汚染イベントでも、数万ポンド相当の生産ランを危険にさらす可能性があり、高品質な入力を確保するための信頼できる調達層の必要性を強調しています[13][14].
よくある質問
クリーンルームにおける資格とバリデーションの違いは何ですか?
資格とバリデーションは、クリーンルームのコンプライアンスを維持する上で異なるが同様に重要な役割を果たします。
資格は、クリーンルームとそのシステムが適切に設置され、意図した通りに機能することを確認することに関するものです。このプロセスには、設計資格 (DQ), 設置資格 (IQ), および運用資格 (OQ). が含まれます。各ステップは、クリーンルームが設計仕様を満たし、効果的に運用されていることを確認します。
バリデーション, 一方で、実際の生産中に必要な環境を一貫して提供するクリーンルームの能力に焦点を当てています。これは、長期的な信頼性、安全性、および規制基準への準拠を確保することに関するものです。
培養肉エリアに適したISOクラス/GMPグレードの選び方は?
培養肉の生産に適したISOクラスまたはGMPグレードを選ぶ際には、生産の特定の段階と関連する汚染リスクに依存します。
- ISOクラス5: 無菌状態を維持することが重要な初期培養段階に最適です。
- ISOクラス6: 清潔さと実用性のバランスを取るバイオリアクター操作に理想的です。
- ISOクラス8: 汚染リスクが低い収穫および移送プロセスに適しています。
無菌状態を妥協できないエリアでは、より高い清潔基準を維持することが不可欠です。さらに、規制要件を満たすためには適切な環境管理が必要です。
どのような変更がクリーンルームの即時再検証を必要としますか?
クリーンルームのレイアウトの変更、新しい機器の追加、または無菌性やコンプライアンスに影響を与える可能性のある環境制御の更新など、大きな変更が行われた場合、即時再検証が必要になります。このような変更は重要な条件に影響を与える可能性があるため、再検証によりすべてが規制要件を引き続き満たしていることを確認します。










