

クリーンルームでは、GMPコンプライアンスが詳細な監視と正確なデータ記録を要求することで、一貫した品質と安全性を確保します。培養肉施設においては、クリーンルームの条件におけるわずかな逸脱でも細胞の成長を損なったり、生産バッチを汚染したりする可能性があるため、特に重要です。
重要なポイント:
- GMP基準: データの完全性に焦点を当て、ALCOA+フレームワーク(帰属可能、判読可能、同時性、オリジナル、正確、完全、一貫、持続、利用可能)に従います。
- 重要なパラメータ: 空気中の粒子、微生物数、温度、湿度、圧力を監視してリスクを早期に検出します。これには、これらの重要なパラメータを維持できる正確なセンサーを選択する必要があります。
- データシステム: 役割ベースのアクセス、監査証跡、電子および紙の記録の安全な保存を備えた検証済みのバイオプロセス制御システムを使用します。
- 一般的なリスク: 手動データ処理のエラー、管理されていない設定変更、不適切な保管方法を避ける。
- 培養肉のためのカスタマイズされたGMP: バイオリアクターの条件や洗浄剤の残留物など、特有のリスクに対応するために監視戦略を調整する。
培養肉の研究開発において、堅牢なデータ管理は製品の安全性、規制の遵守、スケーラブルな運用を保証する。既知の脆弱性に積極的に対処し、後の高額な規制問題を回避する。
クリーンルームデータの完全性に関する主要なGMP要件
ALCOA+原則の理解
GMPデータの完全性の基盤はALCOA+フレームワークにある。MHRA, EMA , およびWHOなどの規制機関は、クリーンルームの記録が信頼できるかどうかを判断するためにこれを使用する。ALCOA+ は以下を表します:Attributable(帰属可能)、Legible(判読可能)、Contemporaneous(同時性)、Original(原本性)、Accurate(正確性)、Complete(完全性)、Consistent(一貫性)、Enduring(持続性)、およびAvailable(利用可能) . これらの用語は、クリーンルームの運用において実用的な重要性を持っています。
- Attributable(帰属可能): すべての記録 - それが粒子数、圧力測定、または清掃ログであれ - は、誰が記録したのか、日付、時間、および関連する機器の詳細を明確に示す必要があります。
- Legible(判読可能): 記録は読みやすく、解読しやすく、レビューや検査の際に明確であることを保証する必要があります。
- Contemporaneous(同時性): データはリアルタイムで記録されなければなりません。遅延または事後の記録は、記録の信頼性を損なう可能性があります。
- Original(原本性): データは、無許可の編集や変更をせずに、最初に取得された形のままであるべきです。
- 正確: 記録された値は、観察された結果を正確に反映し、エラーや操作がないことが求められます。
- 完全: 逸脱や仕様外の結果を含むすべての関連エントリーを文書化する必要があります。
- 一貫性, 持続的, かつ利用可能: 記録は正しい順序に従い、必要な保存期間中は無傷で保存され、レビューや検査のためにすぐにアクセスできる状態であるべきです。
規制当局はこれらの原則を非常に重視しています。例えば、MHRAの検査レビューでは、3年間のGMP不適合声明の80%以上がデータインテグリティの問題に関与していることが明らかになりました[5]. ALCOA+を日常のワークフローに組み込むために、施設はよく構造化されたフォームを採用し、必須フィールドを強制し、定期的な監査トレイルレビューを実施することができます。
ALCOA+を基盤として、次のステップはこれらの原則が紙、電子、およびハイブリッドシステム全体で維持されることを保証することです。
フォーマット全体でのデータインテグリティの確保
培養肉施設では、データがバッチリリースの決定に直接影響を与えるため、すべての記録フォーマットでのインテグリティの維持は不可欠です。GMPは、紙と電子の記録の両方に同じレベルのインテグリティを要求しますが、特定の管理はフォーマットによって異なる場合があります。
- 紙のシステム: ベストプラクティスには、消えないインクを使用した管理されたフォームと明確な訂正プロトコル(e.g. 、署名と日付を伴う一重線での訂正)が含まれます。安全なアーカイブと定義された保存期間の遵守も重要です。
- 電子システム: これらは、Annex 11およびGAMP 5. に準拠した検証済みソフトウェアで動作する必要があります。主な機能には、役割ベースのアクセス、ユニークなユーザー認証、包括的な監査証跡、および同期されたタイムスタンプが含まれます。不規則性を特定し対処するために、定期的な監査証跡のレビューが不可欠です。
- ハイブリッドシステム: これらは電子記録と紙の記録の両方を含むため、最もリスクが高いとされています。例えば、機器が電子データを生成し、それが後で紙のログに転記される場合、元の電子出力は主要な記録として保存されなければなりません。電子記録と紙の記録の間の不一致を検出し解決するために、ワークフローに調整ステップを組み込む必要があります。これは、培養肉の生産において特に重要であり、わずかなデータの不一致でも汚染管理措置. を損なう可能性があります。
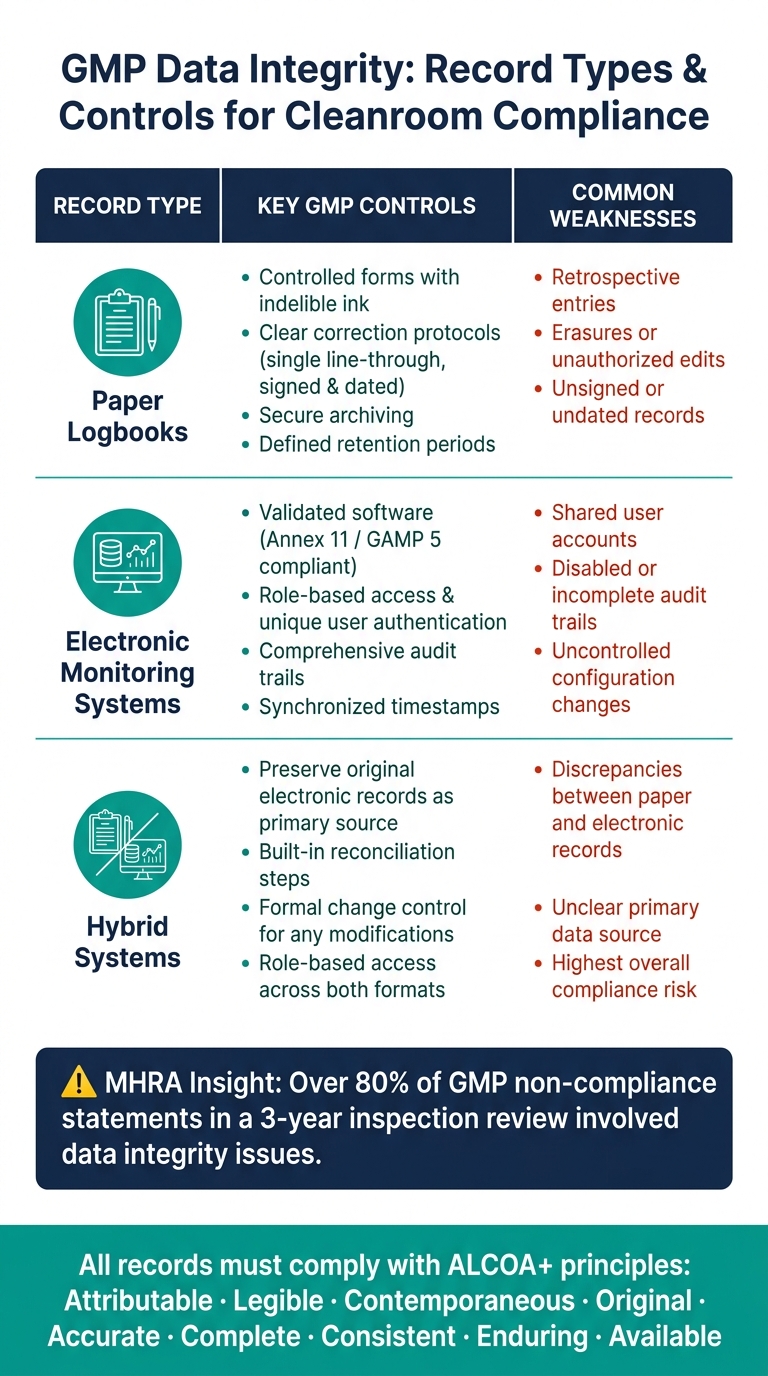
以下の表は、各記録タイプの主要な管理と一般的な弱点をまとめたものです:
| 記録タイプ | 主要なGMP管理 | 一般的な弱点 |
|---|---|---|
| 管理されたフォーム、消えないインク、明確な訂正プロトコル、署名と日付のあるエントリー | 遡及的なエントリー、消去、署名のない記録 | |
| 電子監視システム | 検証済みソフトウェア、役割ベースのアクセス、監査証跡、時間同期 | 共有ユーザーアカウント、無効または不完全な監査証跡 |
| ハイブリッドシステム | 元の電子記録を保持する; 調整手順を実施する | 紙と電子記録の間の不一致、不明確な主要データソース |
コンプライアンスを確保するために、記録はその重要度に応じて分類されるべきです。培養肉施設において、バッチリリースの決定や汚染管理に関連するデータ(e.g. 環境モニタリング結果、HVACアラームログ、またはフィルターの完全性試験データ)は、最も厳しいアクセス制御、頻繁なレビュー、そして堅牢な監査証跡管理の対象とすべきです。
sbb-itb-ffee270
GMPクリーンルーム定期環境モニタリング& 21CFR part 11 データインテグリティ
クリーンルームデータライフサイクルの管理
GMPデータインテグリティ: 記録タイプ& クリーンルームコンプライアンスのための制御
クリーンルームデータライフサイクルの段階
培養肉施設におけるクリーンルームデータライフサイクルは、各段階に特定のコンプライアンス要件があるいくつかの段階を含みます。
データ生成が始まりを示します。これは、粒子カウンター、差圧センサー、温度および湿度プローブ、バイアブルエアサンプラー、表面接触プレート、清掃確認ログなどの機器からの測定値を含みます。各パラメーターについて、サンプリング頻度、指定されたオペレーター、校正された機器が文書化されている必要があります。これらのモニタリング作業を接種、細胞拡大、収穫などの生産段階と一致させることで、環境管理が製品の品質と安全性に直接結びついていることを示すのに役立ちます。
生成されたデータは、キャプチャおよび転送段階に入ります。理想的には、電子システムは、個々のユーザーアカウントに結び付けられたタイムスタンプ付きのエントリで自動的に測定値を記録する必要があります。紙ベースのエントリの場合、データは消えないインクを使用してリアルタイムで記録され、電子システムにデータを転送する際には照合チェックが行われる必要があります。
保管段階も同様に重要です。生データと処理済みデータの両方を保存する必要があります。これにより、報告された値を元の記録に遡ることができます。これには、役割ベースのアクセス制御と定期的なバックアップテストを備えた安全で検証済みのリポジトリが必要です。バックアップは、プライマリシステムとは別の場所に保存し、必要に応じて復元できることを確認するために定期的に検証する必要があります。
最後に、アーカイブがライフサイクルを完結させます。記録は、もはや積極的に使用されていないが、必要な保存期間中に取得可能でなければならない場合、読み取り専用の制御アクセス状態に移行します。培養肉施設では、開発段階のデータをアーカイブすることで、将来の検証作業をサポートすることもできます。
これらの段階を明確に理解することは、以下に示すように、リスクを効果的に管理するために不可欠です。
データ管理における一般的なリスク
転送中のデータ処理は、かなりのリスクを伴います。手動の転記エラーや遡及的な入力は、データの整合性を損なう可能性があります。これを避けるために、すべての入力はリアルタイムでALCOA+原則(帰属可能、判読可能、同時性、オリジナル、正確性、さらに完全性、一貫性、持続性、利用可能性)に従う必要があります。
構成変更もまた大きな懸念事項です。正式な変更管理なしにアラーム限界、センサーのマッピング、またはシステムクロック設定を調整すると、変更前後に記録されたデータの信頼性が損なわれる可能性があります。さらに、ストレージの障害 - データベースの破損、未テストのバックアップ、環境要因による紙のアーカイブの損傷など - により、重要な記録がアクセス不能になることがあります。これらのリスクを軽減するために、すべてのデータストリームが明確な所有権を持つ指定されたアーカイブポイントにマッピングされ、規制検査中に脆弱性が指摘される可能性を減らします。
クリーンルーム監視システムのためのGMP管理
監視システムのための重要な管理
GMP基準を満たす監視システムは、校正されたセンサー、安全なデータ処理、効果的なアラーム管理に依存しています。温度、相対湿度、差圧、非生存粒子数、生存微生物サンプリングなどのパラメータのセンサーは、文書化されたスケジュールに従って校正され、認識された基準に追跡可能でなければなりません。これらのセンサーからのデータ転送を自動化し、同期されたタイムスタンプを含めることで、手動エラーのリスクを最小限に抑えます。
アラーム管理も同様に重要です。アラームの限界は、ISO 14644-1 クラス限界や EU GMP 附属書1 ガイダンスなどの規制フレームワークに一致させる必要があります。トリガーされたすべてのアラームには、ユーザーの詳細、タイムスタンプ、およびコメントを含む記録された応答が伴わなければなりません。アラーム応答を記録しないことは、コンプライアンスの脆弱性を生み出します。
システム全体で役割に基づくアクセス制御を厳格に実施する必要があります。アラーム限界、センサー構成、またはシステムクロック設定の変更には管理者レベルの承認が必要であり、これらの変更は正式な変更管理プロセスに従わなければなりません。構成の更新、データの削除、電子署名、センサーの調整など、GMP関連のすべてのアクションには監査証跡が必須です。これらの証跡は、MHRAデータインテグリティガイダンスおよびEU GMP附属書11に記載されているように、定期的にレビューする必要があります。
培養肉生産システム, では、これらの制御が特に重要です。環境条件が細胞の生存率に直接影響を与えるためです。
これらの制御が整ったら、システムを検証し、変更を慎重に管理して、継続的なコンプライアンスを確保する必要があります。
バリデーションと変更管理手順
監視システムは、センサーの精度、アラーム機能、データの完全性、バックアッププロセス、監査証跡を検証するために、IQ、OQ、PQの段階を通じてバリデーションを受ける必要があります。あるいは、GAMP 5の原則とAnnex 11に沿ったライフサイクルアプローチを使用することもできます。
EU GMP Annex 1(2022年改訂版)は、環境監視システムが「適切に資格を持ち、バリデーションされている」ことを要求し、電子記録がAnnex 11の基準を満たすことを義務付けています。これらの要件は、GMP準拠の施設の基準を設定します。
データの完全性、アラーム機能、または追跡可能性に影響を与える可能性のある変更は、正式な変更管理プロセスを経る必要があります。ソフトウェアパッチのような一見些細な更新であっても、監査証跡を乱す可能性があるため、事前の影響評価なしに実施すべきではありません。
異なる種類のデータには、正確でタイムリーな報告を確保するために、特別なGMP管理が必要です。
データタイプとコンプライアンス要件の比較
クリーンルームモニタリングにおける各データタイプには、GMPコンプライアンスを維持するための特定の要件があります。以下の表は、さまざまなデータタイプの主要な管理を示しています:
| データタイプ | 監視モード | 主要GMP管理 | 制限基準 |
|---|---|---|---|
| 非生存粒子数 | グレードA/Bでの操作中は連続または頻繁に、他のグレードでは定期的に | 検証済みの粒子カウンター、自動データキャプチャ、アラーム付き、標準にトレーサブルな校正、構成変更の監査証跡 | ISO 14644-1 クラス制限、グレードA/Bゾーンのための附属書1ガイダンス |
| 差圧 | 連続、アラーム付き | 校正済みの圧力トランスミッター、自動のタイムスタンプ付き記録、アラーム確認のログ、ゾーン間で10–15 Paの差圧を維持 | 附属書1、施設固有の部屋分類設計 |
| 温度と相対湿度 | 重要なプロセスでは連続的に、それ以外では定期的に | 校正済みプローブ、自動データキャプチャ、トレンド分析、プロセスおよび規制のニーズに基づくアラーム限界 | プロセス知識、規制ガイダンス、製品の感受性 |
| 生存可能な空中微生物 | 断続的(アクティブエアサンプリング)、重要な操作では頻度を増加 | 認定サンプラー、制御されたサンプリング手順、ラボへのチェーンオブカストディ、バッチと場所にリンクされた結果、調査準備済みの記録 | EU GMP 附属書1のグレード別微生物限界 |
| 表面接触結果 | 定期的、清掃後および操作後 | 制御されたサンプリング方法; ラボのトレーサビリティ; グレード固有の制限に対してレビューされた結果; 清掃記録にリンク | EU GMP 附属書 1; 施設のSOP |
各データタイプには、定義された受け入れ基準、定期的なレビュー スケジュール、保持ポリシー、および逸脱に対するエスカレーション プロセスが必要です。すべてのデータタイプに対して均一なレビュー基準を適用することは、規制当局がますます精査している一般的な誤りです。各データタイプの特定のニーズに合わせてレビュープロセスを調整することで、コンプライアンスと運用効率を確保します。
報告、レビュー、および是正措置
コンプライアントな報告書の作成
ALCOA+基準に沿った報告書は、監査のために徹底的で正確かつアクセス可能でなければなりません。GMP準拠のクリーンルームモニタリング報告書は、簡潔で検証可能であり、環境管理を示しながらバッチリリースの決定をサポートできるものであるべきです。最低限、これらの報告書は以下を含むべきです:
- モニタリング期間と範囲をカバーする。
- 計画されたスケジュールと比較したサンプリング活動を要約する。
- 警報または行動限界が超えたかどうかを明確に述べる。
トレンド分析は、これらのレポートの重要な要素であり、管理図、移動平均、100サンプルあたりの逸脱率などの統計ツールを使用して、徐々に変化を特定します。例えば、バイオリアクターの収穫ライン付近での生存可能数の月次トレンドが着実に増加していることは、単一の限界超過イベントよりもはるかに多くの洞察を提供します。メンテナンス活動、プロセス調整、または人員変更などの注釈を追加することで、データの解釈が容易になり、監査対応が向上します。
監査証跡のレビューは、訓練を受けた担当者がその所見を詳細に記録する必要がある、もう一つの重要なステップです。これには、特定のシステムイベントを誰がレビューしたかの記録、異常の記録、フォローアップアクションの詳細を、署名と日付付きの記録に含めることが含まれます。
レポートの頻度は、関連するリスクに合わせる必要があります。例えば:
- バッチ関連のレポートは、各生産ランごとに作成されます。
- 定期的な環境モニタリングの概要は通常、週次または月次で行われます。
- トレンドレポートは、ドリフトの初期兆候を特定するために、月次または四半期ごとに作成されます。
選択された報告頻度は、標準作業手順書(SOP)で正当化され、一貫して遵守されなければなりません。これらのプロトコルは、逸脱が特定された場合に是正措置を開始するための基盤も提供します。
コンプライアンスの失敗への対応
逸脱が発生した場合、構造化され追跡可能な対応が不可欠です。各逸脱には、固有の識別子、明確な説明、および製品への影響とデータの完全性の両方を評価するリスク評価が必要です。逸脱はまた、(軽微、重大、またはクリティカル)に分類され、バッチリリースに影響があるか、追加のテストが必要かを評価するために、特定のバッチまたは生産ロットにリンクされなければなりません。
CAPA(是正および予防措置)フレームワークは、GMPの失敗に対処するための中心的な要素です。効果的なCAPAには、「人的エラー」にイベントを帰する以上のものが必要です。EMAおよびPIC/Sのガイダンスは次の点を強調しています:
「重大な逸脱、OOS結果、データインテグリティ問題を適切に調査しないこと」が執行措置の再発原因である。
5 Whysやフィッシュボーンダイアグラムなどの根本原因分析ツールは、手続き上のギャップ、不十分なトレーニング、技術的なコントロールの弱点に関連するかどうかにかかわらず、システム的な問題を明らかにするために非常に貴重です。是正措置は、データの取得と保存中に特定されたリスクに対処する必要があります。
各CAPAには、測定可能な有効性基準を含める必要があります。例えば、「6か月間、アクションレベルを超えるGrade Bの生存可能な逸脱が繰り返されないこと」。さらに、これらの基準が満たされていることを確認するためのフォローアップレビューが不可欠です。一般的なCAPAメトリクスには以下が含まれます:
- 未完了のアクションの数。
- 完了までの平均時間。
- 期限内に完了したアクションの割合。
- CAPAの有効性を示す強力な指標である再発逸脱の率。
2015年から2019年のGMP警告書のレビューにより、データインテグリティの指摘の65〜70%が不十分な調査、欠落した文書、またはデータの適切なレビューと報告の失敗に起因していることが明らかになりました[2]. これは、適切に管理された施設の証拠として、堅牢な報告と応答性のあるCAPAフレームワークの重要性を強調しています。
培養肉施設におけるGMPコンプライアンスの維持
培養肉の生産における安全性と品質を確保するために、施設はこの新興分野の特定の課題に対応するために確立されたGMP管理を適応させる必要があります。クリーンルームのデータインテグリティは製品の安全性を維持する上で重要な役割を果たすため、培養肉のGMP実践を洗練することが不可欠です。
培養肉のためのGMP実践の調整
EU Annex 1のようなGMPフレームワークは、もともと医薬品のために作成されたものであり、培養肉生産における独自のリスクに対処するために調整が必要です。FMEAやHACCPスタイルの分析のような正式なリスク評価は、生産の各段階にGMP原則を整合させるための堅固な基盤を提供します。細胞バンクの解凍、バイオリアクターの接種、細胞の拡大、収穫といった重要な操作には、Annex 1で指定された適切なクリーンルームの分類、ガウンのプロトコル、環境モニタリングが求められます。一方で、スキャフォールドの取り扱いや包装などの下流工程は、規則 (EC) No 852/2004, の下で、プロセス全体を通じてトレーサビリティとデータの完全性が維持される限り、食品グレードの衛生GMP基準に準拠することができます[6][9][14].
環境モニタリング戦略は、従来の製薬病原体を単にターゲットにするのではなく、培養肉と食品安全に関連する微生物に焦点を当てるべきです。サンプリングは、開放型バイオリアクター、培地調製ゾーン、スキャフォールド取り扱いステーションの近くなど、高リスクエリアを優先する必要があります。これらの場所は、文書化された気流パターンと人員の動きの分析に基づいて選択されるべきです[9][10].
培養肉のバイオリアクターによって生成される大量のデータを考慮すると、システムはこのデータをキャプチャし、タイムスタンプを付け、検証済みのリポジトリに安全に保存できる必要があります。機器からの元の生データファイルは常に主要な記録として識別され、コンプライアンスを確保する必要があります[7][8].
清掃および消毒プロトコルも慎重に検討する必要があります。製薬環境で許容される残留物は、培養肉の生産における細胞の接着や分化を妨げる可能性があります。清掃剤の検証データは、環境モニタリングプログラムの一部として収集および維持されるべきです[3][4].
業界リソースの利用 Cellbase
特殊な調達プラットフォームは、培養肉施設の特定のニーズを満たすために非常に貴重です。GMP対応のクリーンルーム機器は、侵入保護と清掃性の基準を満たし、検証済みのデータシステムとシームレスに統合される必要があります。サプライヤーは、機器に加えて、監査証跡機能、データエクスポート形式、アラーム構成、校正手順を含む詳細な仕様を提供しなければなりません。
システムを
- データインテグリティ機能: 安全な監査証跡、タイムスタンプ付き記録、役割ベースの権限、ユニークなユーザーログイン
- システム互換性: 標準通信プロトコルとAPIのサポートによる集中データストレージ
- 校正とメンテナンス: 包括的なドキュメントの利用可能性
- 適格性サポート: サプライヤー提供のIQ/OQテンプレートによる効率的なバリデーション
- クリーンルーム適合性: 清掃を容易にする材料とデザイン
調達プロセスの早い段階でサプライヤーのドキュメントを要求することで、将来的な適格性の問題を回避するのに役立ちます[3][4].
結論
重要なポイントの要約
クリーンルームのデータ管理におけるGMPコンプライアンスは、プロセス、記録、およびそのデータに基づく意思決定の管理を示すことを中心に展開されます。記録が信頼性を欠いている場合、それが記録するプロセスも同様に疑わしいものとなります。この原則は、環境モニタリングログ、バイオリアクターの出力、逸脱報告、または校正証明書を扱う場合でも普遍的に適用されます。
この議論を通じて、4つの中心的なテーマが浮かび上がりました。第一に、データの完全性, はALCOA+の原則に導かれ、コンプライアントなクリーンルーム文書の基盤です。第二に、ライフサイクル管理は、データが正確に記録され、迅速にレビューされ、安全に保管され、必要な期間保持されることを保証します。第三に、検証済みで変更管理されたモニタリングシステムは、どのSOPも代替できない技術的基盤を形成します。MHRAによる2016年から2021年までのGMP検査の分析で強調されているように、一般的な欠陥には不完全な記録と不十分な監査トレイルのレビューが含まれ続けています[1]. 最後に、正確で追跡可能な報告は、生データがバッチの決定、調査、是正措置にリンクできることを保証し、規制の期待に応えます。
培養肉施設において、これらの原則はさらに重要性を増します。R&Dスタイルのワークフローと生産レベルの管理を組み合わせるという課題は、両方の運用環境を橋渡しするための強力なデータガバナンスを要求します。適切なクリーンルームデータ管理は、一貫性と再現性を確保するだけでなく、規模拡大の準備を整え、規制当局、投資家、消費者に製品の安全性を示します。
最も実行可能なアドバイスは?監査人が指摘する前に既知のリスクに対処することです。ハイブリッドの紙–電子システム、共有ユーザーログイン、データレビューの遅延、制御されていないローカルストレージなどの脆弱性は予測可能で防止可能です。これらの問題を積極的に解決することは、品質事故後にデータの痕跡を再構築するよりもはるかに効果的で、コストも低く抑えられます。
これらのニーズに合わせた監視機器、センサー、またはインフラストラクチャを求めるチームには、
よくある質問
日々のクリーンルーム記録でALCOA+をどのように示すことができますか?
日々のクリーンルーム記録にALCOA+の原則を適用するには、以下を確認してください。
- 帰属可能 : 各エントリのタイムスタンプを含め、責任者を明確に特定します。
- 判読可能: 記録は読みやすく、曖昧さがないことが必要です。
- 同時性 : 活動が行われた時点で情報を記録します。
- オリジナル: データの最初の記録を保持し、コピーや転写を避けます。
- 正確: すべてのエントリーが誤りなく真実のデータを反映していることを確認します。
- 完全: 関連するすべてのデータとメタデータを漏れなく含めます。
- 一貫性: 記録に論理的で連続した順序を維持します。
- 耐久性: 長期保存に適した形式と材料を使用します。
- 利用可能: 必要に応じてレビューや監査のために記録をアクセス可能にしておきます。
これらのステップは、クリーンルームデータ管理におけるGood Manufacturing Practice (GMP)の遵守を確保するために重要です。
ハイブリッド紙–電子システムにおける主なデータ整合性リスクは何ですか?
情報を複数の場所に保存することは、データの正確性を検証する際に複雑さをもたらします。さらに、手動でのデータ入力は人為的なエラーのリスクを増大させ、管理が不十分なシステムやスタンドアロンシステムは、記録が改ざんや削除に対して脆弱になります。これらの問題は、コンプライアンスを維持しデータの整合性を保つために、強力なデータ管理の実践が必要であることを強調しています。
監査官は、システムのバリデーションと変更管理の監視にどのような証拠を期待していますか?
監査官はしばしば、システムのバリデーションを示す文書化された証拠を求めます。これには、以下のような重要なパラメータのテストが含まれます:
- HEPAフィルターの整合性: フィルターが要求される性能基準を満たしていることを確認する。
- 気流と圧力差: 制御された環境を維持するために、これらが許容範囲内であることを確認します。
- 環境モニタリングデータ: 施設が清潔さと汚染管理の要件を満たしていることを示します。
検証試験を超えて、変更管理活動の記録を維持することも同様に重要です。これは、フィルター交換や施設の改造, などの行動をカバーし、システムが期待通りに機能し続け、規制基準を遵守していることを証明するのに役立ちます。










