

成長媒体の検証は、英国市場での承認を求める培養肉企業にとって必須のステップです。このプロセスは、英国新規食品規制 (EU 2015/2283)などの厳格な規制枠組みの下で、製品の安全性、品質、およびコンプライアンスを保証します。以下は知っておくべきことです:
- 主な要件: 成長媒体は、毒性学、汚染管理、栄養品質、およびアレルギー性の基準を満たす必要があります。
- 英国の規制: 食品基準庁(FSA)は、HACCP原則への準拠と動物由来製品(POAO)としての分類を要求しています。
- 世界基準: 英国とEUは類似の枠組みを共有していますが、米国はFD&C法の下でCGMP規制に従っています。
- 検証プロセス: 組成、純度、機能性、サプライヤーのコンプライアンスの徹底的なテストと、堅牢な文書化を含みます。
- 支援イニシアチブ: 2025年に開始された英国の規制サンドボックスは、企業がこれらの基準を満たすのを支援します。
適切な検証は安全性を確保し、信頼を築き、法的要件に沿ったものとなります。この記事では、テスト方法、サプライヤーの資格、規制提出のヒントを含むステップバイステップのプロセスを詳しく説明しています。
成長媒体の規制基準
基準とガイドライン
培養肉生産の重要な要素である成長媒体は、厳格な国際規制基準を満たさなければなりません。これらの基準は地域ごとに異なり、組成、安全性、純度に関する特定の要件があります。
英国, では、成長媒体は新規食品規則(統合規則(EU)2015/2283)に基づいて規制されています。市場で承認される前に、徹底的な安全性評価が必要です[1]. 食品基準庁(FSA)は、細胞培養製品を規則(EC)853/2004の下で動物由来製品(POAO)として分類しています。この分類により、生産者は危害分析重要管理点(HACCP)原則に基づいた食品安全管理システムを実施することが義務付けられています[3]. FSAはまた、成長培地の組成に関する詳細な技術ガイダンスを開発中であり、さらなる更新が期待されています[1]. これらの枠組みは、より具体的な規制要件の基盤を提供します。
アメリカ合衆国, ではアプローチが異なります。成長培地の成分は、連邦食品・医薬品・化粧品法(FD&C法)第501条(a)(4)(B)に記載された現行の適正製造基準(CGMP)要件を満たす必要があります[4]. FDAは培地成分を「供給品および試薬」として分類しており、21 CFRパート210および211によって管理されています。これらのコンポーネントは、汚染を防ぐために品質検証を受ける必要があります[4]. 興味深いことに、培養肉メディアの合成成分 - アミノ酸、ビタミン、塩類など - は、21 CFR 864.2220の下でクラスI医療機器として分類されることが多く、事前市場通知要件から免除されています [6][7].
欧州連合, では、規制の枠組みは英国と密接に一致しており、EU規則2015/2283に従っています。&欧州食品安全機関(EFSA)が認可プロセスを監督しています[1]. ICH Q6Bガイドラインによれば、抗生物質、誘導体、その他の成分を含む成長メディアの成分は、プロセス関連不純物として扱われます。これらの不純物は管理され、許容レベルまで減少させる必要があります [5]. 可能な場合、賦形剤および試薬は薬局方の基準に準拠する必要があります[5].
| 管轄区域 | 主要規制 | 分類 | 安全システム | メディア監視 |
|---|---|---|---|---|
| イギリス (GB) | 同化規制 (EU) 2015/2283 [1] | 動物由来製品 (POAO) [3] | HACCP (規則 852/2004) [3] | FSA/FSS サンドボックスガイダンス [1] |
| 欧州連合 / NI | 規則 (EU) 2015/2283 [1] | 動物由来製品 (POAO) [3] | HACCP (規則 852/2004) [3] | EFSA 認可プロセス [1] |
| アメリカ合衆国 | FD&C法第501条(a)(4)(B) [4] | 新動物用医薬品 / 食品 [4] | CGMP (21 CFR 210/211) [4] | FDA CVM / USDA-FSIS [4] |
培養肉の規制要件
培養肉の生産者は、成長媒体のすべてのバッチが厳格な安全性と品質基準を遵守していることを確認しなければなりません。成長培地の検証は、これらの製品に対する広範な規制枠組みの重要な側面です。HACCP原則(規則(EC)852/2004)に基づき、成長培地は主要な投入物および汚染の潜在的な原因として特定されています - 化学的、微生物的、またはその他の[3] . FSAはこの懸念を強調しています:
"細胞培養製品の生産における主な危険は、細胞株の同一性(および一貫性)、生産プロセス中に導入される危険(微生物汚染、成長培地および最終製品中の残留成分)、およびアレルゲンに関するものです。" [3]
成長培地の配合に変更がある場合、直ちにHACCPの見直しが必要です[3]. 英国では、フローダイアグラムの正確性と管理措置の有効性を確保するために、実施前に検証を行う必要があります[3].
アメリカ合衆国では、FDAはすべての試薬と培地成分が有害な物質を持ち込まないように厳格な品質基準を満たすことを義務付けています[4]. サプライヤーおよび契約ラボはCGMP規制を遵守しなければならず、遵守しないサプライヤーは「不良品」と分類されるのを防ぐために排除されるべきです[4]. FDAはこれの重要性を強調しています:
「すべての新しい動物用医薬品、ACTPを含む、は安全性に関する連邦食品・医薬品・化粧品法(FD&C法)の要件を満たすためにCGMPに従って製造されなければなりません。" [4]
現在、BlueNalu, Gourmey, Hoxton Farms, Mosa Meat,
成長媒体の検証手順
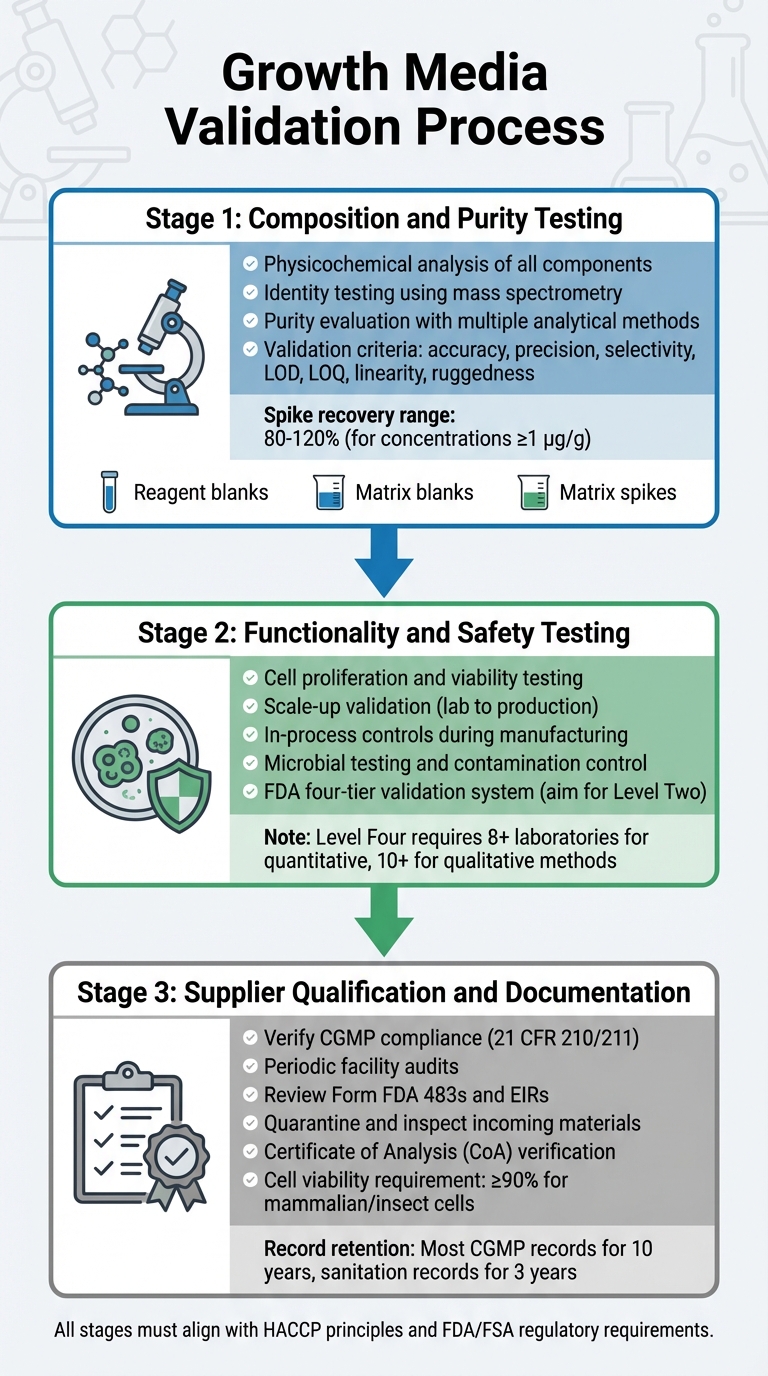
培養肉の規制承認のための成長媒体検証プロセス
成長媒体の検証は、その組成、機能、安全性、およびサプライヤーのコンプライアンスを調査する詳細なプロセスを含みます。 各ステップは前のステップに基づいて構築され、規制要件に沿った堅牢な検証プロセスを確保します。これには、組成、機能、およびバイオプロセスメディアサプライヤーのコンプライアンスのテストが含まれます。
組成と純度のテスト
最初の段階では、各成分の徹底的な物理化学的分析に焦点を当てます。これには、アミノ酸、ビタミン、無機塩などの成分の正確な組成、物理的特性、および分子構造の特定が含まれます [5]. 分子構造を確認するために、同定試験は物理化学的、生物学的、および免疫化学的技術を含む高度に特異的な方法を使用します。質量分析法などのツールを使用して、分子の断片化パターンを通じて分子の同一性を検証します[8].
純度評価には、目的の成分を不純物から分離するための複数の分析方法が必要です。これらの試験は、プロセス関連および製品関連の不純物の両方に対処する必要があります[5]. 分析技術は、正確性、精度、選択性、検出限界(LOD)、定量限界(LOQ)、直線性、および堅牢性を含む厳格なバリデーション基準を満たす必要があります[8]. バリデーションプロトコルには、次のものも含める必要があります:
- 試薬ブランクは、試薬が分析物から自由であることを確認します。
- マトリックスブランクは、サンプル環境が干渉しないことを確認します。
- マトリックススパイクは、回収率と正確性を推定します。
1 µg/g(ppm)の濃度での定量法の場合、許容されるスパイク回収率は通常80%から120%の範囲です[8] .
一貫性を維持するために、製造業者は生産を代表するロットから派生した社内の一次基準物質を確立する必要があります。これらは、作業基準物質を校正するための追跡可能な標準として機能します。[5]. 純度試験が完了したら、培地は効果的な細胞増殖をサポートし、安全基準を満たす能力を示さなければなりません。
機能性および安全性試験
組成を確認した後、培地は培養肉の生産をサポートする効果を証明しなければなりません。これには、細胞が増殖し、生命力を維持し、実験室条件から生産量にスケールアップできることを示すことが含まれます。この移行には、必要な規制データを生成するためにパイロットスケールシステムがしばしば必要です。FDAは、製品の一貫性と安全性を確保するために、細胞の継代や収穫のような初期段階から製造中のプロセス管理を要求しています [4].
安全性の検証には、FDAの事前市場評価で概説されているように、厳格な微生物試験と汚染管理が含まれます [9].
FDAは、化学的方法の検証に四段階のシステムを使用しており、レベル1(緊急または限定使用)からレベル4(AOAC/ISO基準を満たす完全な共同研究)まであります [8]. 定期的な規制試験のためには、レベル2の単一ラボ検証, を目指し、包括的な性能評価を含みます [8]. 定量的方法の完全な共同研究には少なくとも8つのラボの参加が必要であり、定性的な方法には10が必要です [8]. メディアのパフォーマンスが検証されたら、すべての原材料が適合したサプライヤーから供給されていることを確認することが重要です。
サプライヤーの資格と文書化
製造業者は、検証済みのCGMP準拠のサプライヤーと協力しなければなりません。サプライヤーは、21 CFR 210/211で概説されている基準を満たす必要があります[4]. 検証には、品質プログラム、手順、および全体的なCGMP準拠を評価するためのサプライヤー施設の定期的な監査が含まれます[4].
契約を結ぶ前に、サプライヤーのコンプライアンス履歴を確認し、Form FDA 483およびEstablishment Inspection Reports (EIRs)を含めます[4]. FDAはこの義務を強調しています:
「他の施設と契約、合意、またはその他の取り決めを結ぶ前に、その施設が適用される規制CGMPに準拠していることを確認する必要があります。」[4]
すべての入荷材料は、放出前に隔離され、検査され、マスター仕様を満たしていることを確認する必要があります[10]. サプライヤーは、分析証明書(CoA)または追跡可能なCGMP/GLP準拠の試験結果を提供する必要があります[10]. 安定した細胞株の場合、文書には追跡可能なクローン履歴が含まれている必要があります[10]. 哺乳類または昆虫細胞は、通常、CGMPプロジェクトでの受け入れには少なくとも90%の生存率が必要です[10]. 記録は規制ガイドラインに従って保持されるべきです[4].
契約はCGMPの責任を明確にし、供給者がテストキットや方法論の変更を提案した場合に製造者に通知することを要求しなければなりません[4]. テストが外部委託される場合、契約ラボが検証済みの分析方法を使用し、FDAに登録されていることを確認してください[4].
規制提出書類の準備
成長培地が検証されたら、次のステップはFDAおよびUSDA-FSISが要求するすべての安全性と品質基準に準拠していることを示す書類を作成することです。この書類は、検証と規制遵守の間の重要なリンクとして機能し、当局に培地の安全性と生産プロセスを明確に示します。
提出書類の必須要素
提出書類には、すべてのアミノ酸、ビタミン、無機塩類、成長因子を一覧にした培地の詳細な内訳を含める必要があります。FDAのガイドラインは、培地自体だけでなく、全体の生産ワークフローを評価することを強調しています。これには、一次および不死化細胞株とバンクの確立、製造管理の実施、すべての成分と投入物の検証が含まれます[11].
さらに、提出書類には、培養材料とそのすべての投入物の食品安全性を証明するための徹底した安全性および毒性評価を含める必要があります。製造管理記録、プロセス検証データ、品質プログラムの文書を含めて、製造が一貫しており、汚染物質がないことを示してください。
メディアで使用されるすべての材料について、社内で準備されたものを含む、供給品および試薬の検証記録を提供する必要があります。USDA-FSISによって規制されている製品については、HACCPプランと衛生プロトコルを含めてください。FDAは、ほとんどのCGMP記録を少なくとも10年間保持することを推奨しており、施設の清掃および衛生記録は最低3年間保持する必要があります[4]. これは、サプライヤーの資格確認の取り組みと一致し、すべての入力がCGMPおよび規制要件を満たしていることを保証します。
施設のコンプライアンスの文書化
人間の消費のために培養肉を生産、加工、または保管する前に、施設はFDAに登録する必要があります[12]. あなたの文書には、包括的な食品安全計画が含まれている必要があります。これには、危害分析(生物学的、化学的、物理的), 予防管理(衛生管理、アレルゲン管理、サプライチェーン対策など)、および監視手順が含まれます[12].
メディアフィルシミュレーションも重要な要件です。これには、無菌操作を確認するための14日間の培養と成長促進試験が含まれます。FDAは次のように説明しています:
「メディアフィルは、重要な(無菌)機器の無菌組立と操作を評価し、オペレーターの資格と技術を評価し、環境管理が適切であることを示す必要があります」[2].
あなたの記録には、供給業者の資格データを含める必要があります。これは、供給業者からの最初の3バッチの培地に対して行われた試験が分析証明書と一致することを確認するためのものです。その他の重要な記録には、環境管理ログ、機器の校正スケジュール、温度監視データが含まれます。USDA規制のプロセスにおいては、HACCPプラン、書面による衛生標準作業手順(SSOP)、リコール手順を準備してください [12][13].
sbb-itb-ffee270
規制に準拠した成長媒体調達のための Cellbase
培養肉のための認定サプライヤー
成長媒体の処方を検証したら、次のステップは規制基準を満たす成分の調達です。これは一般的なサプライヤーから注文するほど簡単ではありません。細胞培養製品には厳しい衛生規制が適用され、すべての成長媒体成分には規制承認のための特定の文書が必要です [3]. そこで
調達機能
このプラットフォームは、透明性のある価格設定と直接メッセージ機能も提供しており、チームが見積もり、分析証明書、その他の規制文書を迅速にリクエストできるようにします。これらの重要な調達機能を、培養肉生産に特化した一つのシステムに統合することで、
結論
規制承認のための成長培地の検証は、単なるチェック項目ではなく、英国市場に培養肉製品を導入するための法的要件です。これには、組成と純度の徹底的なテスト、強力なHACCPプランの実施、そして各段階での詳細な文書化が含まれます。
「食品は安全でない場合、市場に出してはなりません。これは、健康に害を及ぼさず、人間の消費に適さないことを意味します。" - 食品基準庁 [3]
英国食品基準庁の規制サンドボックスは、成長媒体の組成に関する明確な技術ガイダンスを確立するために業界関係者と協力することへのコミットメントを示しています [1]. 適切な検証を優先する企業は、これらのガイドラインが完全に定義されたときにより強い立場に立つことができます。
コンプライアンス基準を満たすことは、単に規制のチェックボックスを埋めることではなく、消費者の信頼を得て製品の安全性を確保することです。厳格な品質テストは、規制承認と市場受け入れの両方の中心にあります。承認プロセスを合理化するためには、強力な検証プロトコルを構築し、正確な記録を維持し、信頼できるサプライヤー. と提携することに焦点を当ててください。これらのステップは、承認を簡素化するだけでなく、消費者の信頼を高める道を開くでしょう。
よくある質問
規制当局の承認を得るための培地の主な検証ステップは何ですか?
規制当局の承認を得るための培地の検証は、製剤が安全で信頼性があり、培養肉の生産に適していることを証明することに関するものです。通常、プロセスは次のようになります:
- リスク評価: 使用する細胞株、製品の目標、および重要な品質属性(pHや栄養成分など)を定義することから始めます。微生物汚染などの潜在的な危険を特定し、これらのリスクを管理するための対策を策定します。
- 試験と仕様: 無菌性、純度、効力などの要因に対する明確な受け入れ基準を設定します。これらの基準が一貫して満たされることを確認するために、確立された試験方法を使用します。
- バリデーション研究: 結果が再現可能で一貫していることを確認するために、設備の適格性評価や複数バッチのテストを含む徹底的なプロセスバリデーションを実施します。
- 安定性試験: 適切な保管条件(通常2–8°C)での意図された保存期間中にメディアの品質を評価することで、時間の経過に伴うメディアの耐久性を確認します。
- 文書化: すべてのテスト結果と分析を含む包括的なバリデーションドシエを作成し、規制要件を満たすようにします。
これらのステップを慎重に対処することで、培養肉生産に必要な安全性と品質基準を満たしていることを示すために必要な証拠を収集できます。
培養肉に使用される培地に関する英国と米国の規制の主な違いは何ですか?
イギリスでは、培養肉の培地の規制は新規食品規則(EU規則2015/2283), に基づいており、これはGB法に保持されています。1997年5月15日以前に一般的に消費されていなかった製品に使用される培地は、食品基準庁(FSA)による正式な新規食品評価を受ける必要があります。このプロセスには、培地の組成、起源、純度に関する詳細な文書の提出が必要です。さらに、HACCPに基づくリスク評価が必要であり、細胞培養プロセス中の汚染物質の管理方法を示す必要があります。
2025年12月以降、FSAは細胞培養製品サンドボックス. を実施しています。このイニシアチブは、新規食品申請のためのガイダンスを提供し、データ収集を迅速化するサポートを提供します。最終承認を得るために、企業はメディアの安全性、一貫性、製造の検証に関する包括的な書類を提出しなければなりません。この承認を得て初めて、製品はグレートブリテンで販売することができます。
対照的に、アメリカ合衆国には成長メディアに特化した新規食品の枠組みがないため、直接的な規制の比較は困難です。英国に拠点を置く企業にとって、これらの厳しい基準にすでに準拠しているメディアコンポーネントを調達することは、承認プロセスを簡素化することができます。
英国の規制サンドボックスは、成長メディアの検証をどのようにサポートしていますか?
英国の培養製品のための規制サンドボックスは、企業が成長メディアの配合をテストし、改良するための整然とした環境を提供します。食品基準庁(FSA)とスコットランド食品基準庁(FSS)によって監督されているこのプログラムは、6か月のフェーズで実施されます。この期間中、企業は安全性試験を実施し、リスク評価を行い、文書を見直しながら、規制当局から貴重なフィードバックを受け取ることができます。
この実践的なアプローチにより、実際の試行と段階的な改善が可能になり、安全データの収集が迅速化され、企業が規制要件に適合するのを助けます。培養肉に取り組んでいる方々にとって、









