

Catatan batch sangat penting untuk kepatuhan dan keamanan produk. Mereka mendokumentasikan setiap langkah produksi, memastikan standar regulasi terpenuhi. Bagi produsen daging budidaya, mempertahankan kesterilan dan catatan terperinci adalah hal yang tidak bisa dinegosiasikan. Inspeksi FDA sering menyoroti masalah seperti data yang hilang, tinjauan yang tidak lengkap, dan tindakan korektif yang buruk, yang dapat menyebabkan peringatan atau gangguan.
Poin Penting:
- Catatan Batch: Dua jenis - Master Batch Record (MBR) (sebagai "resep") dan Batch Production Record (BPR) (sebagai "pelaksanaan").
- Masalah Umum: Kesalahan manusia (50% dari masalah), pemeriksaan dalam proses yang hilang, tinjauan yang tidak lengkap, dan sistem CAPA (Tindakan Korektif dan Pencegahan) yang buruk.
- Standar FDA: Kepatuhan terhadap prinsip ALCOA+ (Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, Available) adalah wajib.
- Solusi: Audit independen, catatan batch elektronik, dan verifikasi pemasok yang ketat dapat meminimalkan kesalahan dan meningkatkan kepatuhan.
Perusahaan daging budidaya seperti UPSIDE Foods telah menetapkan tolok ukur dengan memastikan dokumentasi yang terperinci, keterlacakan input, dan tindakan korektif yang cepat. Dengan belajar dari praktik ini, produsen dapat menghindari jebakan regulasi dan mempertahankan standar kualitas tinggi.
Panduan Komprehensif untuk Dokumentasi dan Pencatatan untuk Kepatuhan FDA dalam Ilmu Hayati
sbb-itb-ffee270
Masalah Umum dalam Dokumentasi Catatan Batch
Laporan inspeksi FDA secara konsisten menyoroti masalah yang berulang: penyimpangan tinjauan catatan produksi termasuk di antara kekurangan GMP teratas yang dikutip oleh regulator [7]. Untuk produsen daging budidaya, kekurangan ini melampaui kesalahan administratif semata - mereka membahayakan kemampuan untuk menunjukkan kondisi steril yang berkelanjutan. Masalah-masalah ini muncul dalam beberapa bentuk, seperti yang diilustrasikan dalam contoh-contoh di bawah ini.
Ulasan Tidak Lengkap dan Ketidaksesuaian
Salah satu masalah umum adalah kegagalan Unit Kontrol Kualitas untuk meninjau catatan batch secara menyeluruh. Alih-alih menjadi bagian integral dari proses pelepasan, ulasan sering terjadi secara reaktif - hanya setelah masalah produk sudah muncul [7]. Pendekatan ini meninggalkan celah signifikan dalam catatan produksi.
Misalnya, Davis City Pharmacy menerima pengamatan FDA 483 karena catatan batch yang kehilangan detail penting seperti jumlah komponen, langkah operasional, dan inisial personel. Demikian pula, CAPS dikutip karena kurangnya tanda tangan yang diperlukan dan verifikasi peninjau dalam entri kunci [3]. Pelanggaran ini bukanlah insiden yang terisolasi; studi menunjukkan bahwa sekitar 52% pelanggaran dokumentasi meningkat ketika sistem manajemen bioproses yang kuat tidak ada [3] .
"Bukan kompleksitas proses yang memicu kutipan - melainkan ketidakkonsistenan, ketidaklengkapan, dan pengawasan yang buruk." - GXP Auditing & Consulting Services [5]
Catatan Pemeriksaan Dalam Proses yang Hilang
Defisiensi lain yang sering terjadi adalah tidak adanya dokumentasi yang tepat untuk pemeriksaan dalam proses. Catatan ini sangat penting, terutama pada titik kontrol kritis dalam operasi aseptik. Misalnya, Nephron Sterile Compounding Center menghadapi kutipan karena gagal mendokumentasikan langkah-langkah berpakaian penting dan prosedur aseptik dalam catatan batch mereka [3] . Untuk produsen daging budidaya, di mana kemandulan sangat penting, kelalaian semacam itu membuat tidak mungkin untuk mengonfirmasi kepatuhan terhadap langkah-langkah pengendalian kontaminasi.
Amphastar juga ditandai karena gagal menyelidiki atau mendokumentasikan varians hasil yang tidak terduga atau ketidaksesuaian output [3]. Risiko dari kelalaian semacam itu sangat jelas. Dalam satu kasus, sebuah fasilitas farmasi yang tidak dikenal pada tahun 2024/2025 ditemukan menyimpan catatan batch yang telah selesai di rak terbuka dan meja. Penyelidik FDA menemukan halaman yang hilang, termasuk tujuh dari satu catatan, dan seluruh bagian "Synthesis Solution" tidak ada dari catatan lain [6].
Kegagalan Verifikasi CAPA dan Supplier GMP
Di luar kesalahan dokumentasi, ketiadaan proses korektif yang efektif dan verifikasi pemasok lebih lanjut merusak keandalan catatan batch.Ketika terjadi penyimpangan produksi tanpa laporan Tindakan Korektif dan Pencegahan (CAPA) yang sesuai, integritas catatan batch terganggu [7]. Misalnya, Eugia Pharma Specialities Limited, yang diperiksa antara 22 Januari dan 2 Februari 2024, menerima FDA 483 karena gagal meninjau ketidaksesuaian dengan memadai. Sistem CAPA mereka yang tidak efektif dan investigasi yang tidak lengkap menyebabkan masalah produksi berulang, memaksa perombakan total prosedur investigasi dan CAPA mereka [9].
Demikian pula, selama inspeksi dari 26 September hingga 25 Oktober 2023, Stokes Healthcare Inc. menunjukkan manajemen ketidaksesuaian yang buruk. Perusahaan gagal memperluas investigasi ke semua batch yang terkena dampak dan menunda penyelesaian analisis mereka [9].
"Ketidaksesuaian tanpa laporan CAPA atau penyimpangan yang sesuai? Itu adalah kegagalan kepatuhan." - GXP Auditing & Layanan Konsultasi [5]
Masalah verifikasi pemasok menambah lapisan kompleksitas lainnya. Empower Clinic Services LLC disebutkan selama inspeksi dari 18 Juli hingga 5 Agustus 2022 karena prosedur pengendalian kualitas yang tidak memadai, termasuk kualifikasi pemasok yang tidak cukup dan proses investigasi yang buruk [9] . Bagi produsen daging budidaya, yang bergantung pada media pertumbuhan, garis sel, dan input penting lainnya, memastikan kepatuhan GMP pemasok sangat penting untuk menjaga integritas catatan batch.
Persyaratan FDA untuk Catatan Batch
Aturan FDA untuk catatan batch berpusat pada 21 CFR Bagian 117, yang menetapkan dasar untuk keamanan pangan.Ketika berbicara tentang daging budidaya, di mana menjaga kesterilan selama fase kultur sel sangat penting, dokumentasi sering kali harus memenuhi standar yang lebih ketat dari Bagian 111 atau Bagian 211, selain Bagian 117 [10][14]. Ini menekankan betapa pentingnya dokumentasi yang tepat untuk memastikan keamanan dan efektivitas produksi daging budidaya.
Standar Inti untuk Catatan Batch
Setiap batch memerlukan dua dokumen kunci:
- Catatan Batch Utama (MBR): Template yang disetujui yang menguraikan proses produksi.
- Catatan Produksi Batch (BPR): Catatan rinci tentang apa yang sebenarnya terjadi selama pelaksanaan produksi [12][2].
BPR harus mencakup detail seperti nomor batch atau lot, detail peralatan, tanggal pembersihan, pengenal komponen, pengukuran yang tepat, dan perbandingan hasil aktual versus teoritis [10][14].
"Catatan produksi batch harus secara akurat mengikuti catatan manufaktur utama yang sesuai dan Anda harus melakukan setiap langkah dalam produksi batch." – 21 CFR 111.255 [12]
Setiap langkah kritis harus dicatat segera, dengan inisial pelaksana dan verifikator dicatat [10][11]. FDA mengharuskan kepatuhan terhadap prinsip ALCOA(+), yang berarti catatan harus Attributable, Legible, Contemporaneous, Original, and Accurate - serta Complete, Consistent, Enduring, and Available [1].
Jika ada penyimpangan dari Catatan Manufaktur Utama, hal tersebut harus diselidiki secara menyeluruh. Ini termasuk mendokumentasikan masalah, melakukan analisis akar penyebab, dan menerapkan rencana Tindakan Korektif dan Pencegahan (CAPA) [8] [1]. Penilaian awal terhadap penyimpangan harus dicatat dalam waktu 24–48 jam setelah terdeteksi [8]. Untuk fasilitas yang menggunakan sistem elektronik, kepatuhan terhadap 21 CFR Part 11 adalah wajib. Ini termasuk tanda tangan elektronik yang tervalidasi dan jejak audit yang aman dan diberi cap waktu [8] [1].
Prosedur Penyimpanan dan Peninjauan Catatan
Proses penyimpanan dan peninjauan catatan yang tepat sangat penting untuk tetap patuh dan memastikan keamanan produk.Dalam produksi steril, seperti daging yang dibudidayakan, setiap detail dalam catatan batch harus menjalani tinjauan yang teliti. Tim Quality Control (QC) bertanggung jawab untuk meninjau semua catatan batch, memantau hasil, dan menguji data sebelum batch dapat disetujui untuk distribusi [10] [13].
"Semua catatan produksi dan kontrol produk obat harus ditinjau dan disetujui oleh unit kontrol kualitas sebelum batch dirilis atau didistribusikan." – 21 CFR 211.192 [2]
Produsen sering kali bertujuan untuk menyelesaikan 95% tinjauan batch dalam 30 hari setelah produksi [2]. Namun, untuk proses steril yang lebih kompleks yang terlibat dalam daging yang dibudidayakan, tinjauan biasanya memakan waktu 7–10 hari, dengan fasilitas berkinerja tinggi mencapai waktu di bawah 7 hari [2]. Sistem catatan batch elektronik dapat secara signifikan mempercepat tinjauan ini, seperti yang terintegrasi ke dalam sistem produksi daging budidaya, - mengurangi waktu hingga setengah dibandingkan metode berbasis kertas - selama mereka divalidasi untuk memenuhi persyaratan Bagian 11 dan menjaga integritas data [1].
Apa yang Dilakukan dengan Benar oleh Perusahaan Daging Budidaya yang Disetujui FDA
Perusahaan daging budidaya yang disetujui FDA telah menetapkan standar tinggi dengan mengadopsi praktik yang mengatasi tantangan dokumentasi dan memenuhi standar keamanan yang ketat.
Ketika UPSIDE Foods menjadi perusahaan daging budidaya pertama yang lulus konsultasi pra-pasar FDA pada November 2022, mereka menetapkan model untuk industri ini.FDA mengeluarkan surat "tidak ada pertanyaan lebih lanjut" setelah meninjau secara menyeluruh proses produksi mereka, yang mencakup pembentukan garis sel, bank sel, kontrol manufaktur, dan semua komponen serta input [16]. Pencapaian ini menyoroti pentingnya dokumentasi yang terperinci dalam memenuhi persyaratan ketat FDA.
Memenuhi Standar Sterilitas dan Kepatuhan
Pencapaian menonjol UPSIDE Foods adalah pendekatan menyeluruh mereka terhadap keterlacakan input. Setiap komponen produksi didokumentasikan dengan cermat, memastikan rantai akuntabilitas yang jelas dari garis sel awal hingga produk akhir [16]. Tingkat transparansi ini memungkinkan peninjau FDA untuk melacak setiap langkah dari proses manufaktur, mengonfirmasi bahwa semua standar keselamatan secara konsisten terpenuhi.
"Konsultasi pra-pasar FDA dengan perusahaan termasuk evaluasi proses produksi perusahaan dan bahan sel yang dikultur yang dibuat oleh proses produksi, termasuk pembentukan garis sel primer dan diabadikan dan bank sel, kontrol manufaktur, dan semua komponen dan input." – U.S. Food and Drug Administration [16]
Perusahaan sukses lainnya mengikuti dengan menerapkan dokumentasi proses aseptik yang terperinci. Ini termasuk langkah-langkah kritis seperti prosedur berpakaian dan operasi penanganan steril [3]. Tidak seperti kegagalan dokumentasi sebelumnya, perusahaan-perusahaan ini menggunakan sistem tinjauan berjenjang, yang melibatkan pemeriksaan operator, pengawasan produksi, dan tinjauan unit kualitas, untuk menangkap potensi kesalahan sebelum pelepasan batch [15]. Sistem catatan batch elektronik juga memainkan peran penting, menegakkan tanda tangan wajib di setiap tahap dan menjaga jejak audit yang tidak dapat diubah sesuai dengan persyaratan 21 CFR Part 11 [3][2].
Praktik ketat ini secara alami meluas ke cara perusahaan menangani penyimpangan dan kegagalan.
Proses CAPA untuk Kegagalan Batch
Ketika batch gagal memenuhi spesifikasi, perusahaan yang disetujui FDA mengambil tindakan cepat dan sistematis. Proses Tindakan Korektif dan Pencegahan (CAPA) mereka mencakup analisis akar penyebab formal, penilaian dampak, dan tindakan korektif yang didokumentasikan dengan jelas [3]. Setiap penyimpangan dikelola dalam kerangka jaminan kualitas terintegrasi, memastikan bahwa semua masalah diselidiki, dibenarkan, dan didokumentasikan secara menyeluruh sebelum produksi dilanjutkan [2].
Ke depan, integritas data akan menjadi fokus utama tindakan penegakan FDA untuk 2024–2025 [1].
Cara Meningkatkan Praktik Catatan Batch Anda
Memperkuat praktik catatan batch memerlukan dokumentasi yang tepat untuk mengatasi kegagalan umum yang sering diidentifikasi selama inspeksi FDA. Berikut adalah beberapa strategi untuk mengatasi tantangan utama.
Lakukan Audit Catatan Batch Independen
Audit pihak ketiga secara teratur dapat mengungkap masalah yang mungkin terlewatkan oleh tinjauan internal. Mulailah dengan memfokuskan pada sistem kritis seperti Sistem Manajemen Informasi Laboratorium (LIMS), Sistem Eksekusi Manufaktur (MES), dan Perencanaan Sumber Daya Perusahaan (ERP). Prioritaskan dokumen dengan dampak regulasi tinggi, seperti catatan pengujian pelepasan, data stabilitas, dan catatan produksi batch.
Salah satu metode efektif adalah pengujian pengambilan sampel.Secara acak pilih batch terbaru dan rekonstruksi riwayat produksi dan laboratorium mereka. Ini dapat membantu mengidentifikasi data yang hilang, tanda tangan yang tidak lengkap, atau kesenjangan dokumentasi yang dapat menyebabkan kutipan regulasi. Periksa silang jejak audit yang dihasilkan sistem dengan entri manual untuk mengidentifikasi perubahan atau penghapusan yang tidak sah.
Tinjau semua laporan Out-of-Specification (OOS) dan Out-of-Trend (OOT) dari tahun lalu. Evaluasi apakah analisis akar penyebab dilakukan secara menyeluruh dan jika Tindakan Korektif dan Pencegahan (CAPA) diterapkan dengan memadai. Perlu dicatat bahwa masalah dokumentasi menyumbang 21% dari surat peringatan FDA, sementara kesalahan manusia berkontribusi pada 50% masalah catatan batch dalam pembuatan farmasi [2].
"Bukan kompleksitas proses yang memicu kutipan - melainkan ketidakkonsistenan, ketidaklengkapan, dan pengawasan yang buruk." – GXP Auditing & Consulting Services [5]
Simulasikan inspeksi regulasi melalui tinjauan tiruan berkala. Praktik ini membantu tim mengenali ketidakkonsistenan dan potensi masalah integritas data sebelum audit yang sebenarnya. Pastikan semua catatan mengikuti prinsip ALCOA+: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, dan Available.
Setelah integritas dokumentasi kuat, fokus pada verifikasi kualitas semua input produksi.
Uji Semua Input untuk Kontaminasi Mikrobiologis
Pengujian kemandirian dan potensi untuk semua input sangat penting - jangan hanya mengandalkan Sertifikat Analisis (CoAs) dari pemasok. Ini sangat penting bagi produsen daging budidaya, karena kontaminasi dapat membahayakan seluruh batch.
Misalnya, pada Februari 2013, Central Admixture Pharmacy Services menghadapi teguran FDA karena kontrol mikroba yang tidak memadai selama pelepasan batch produk steril. Perusahaan harus memperkenalkan prosedur kontrol mikroba yang terperinci dalam Prosedur Operasi Standar (SOP) [4].
Checkpoint mikroba dalam proses dapat mencegah ketergantungan berlebihan pada pengujian produk akhir. Masukkan checkpoint ini ke dalam SOP pelepasan batch dan pertahankan dokumentasi kontemporer yang ketat. Catat semua hasil tes dan langkah-langkah manufaktur saat terjadi untuk menghindari pencatatan mundur atau entri yang tertunda, yang dapat mengakibatkan teguran FDA.
Simpan file pemasok yang komprehensif, termasuk CoA, laporan audit, perjanjian kualitas, dan riwayat penyimpangan terkait bahan yang masuk.
Memperkuat praktik pencatatan batch lebih lanjut melibatkan penyelarasan proses dengan standar yang telah ditetapkan seperti HACCP dan GCCP.
Menyelaraskan Catatan dengan Standar HACCP dan GCCP
Menggabungkan prinsip-prinsip Hazard Analysis Critical Control Point (HACCP) ke dalam catatan batch memastikan variabel proses kritis dipantau dan didokumentasikan sepanjang produksi. Ini termasuk menetapkan titik pemeriksaan pengujian mikroba dalam proses daripada hanya mengandalkan pengujian tahap akhir.
Bagi produsen daging budidaya, kepatuhan terhadap standar Good Cell Culture Practice (GCCP) sama pentingnya. Catatan batch harus mencakup rincian manipulasi aseptik, prosedur berpakaian, dan pemantauan lingkungan yang terkait dengan kriteria pelepasan batch [3][4]. Langkah-langkah ini membantu menjaga kepatuhan dan memastikan keamanan produk.
Data industri menunjukkan bahwa 52% pelanggaran dokumentasi meningkat ketika perangkat lunak manufaktur batch yang tepat tidak tersedia [3][4]. Sebuah contoh kasus: pada Februari 2023, Nephron Sterile Compounding Centre menerima pengamatan dari FDA karena tidak adanya prosedur kontrol untuk memverifikasi variabel proses kritis sebelum pelepasan batch [4]. Ini menyoroti kebutuhan akan dokumentasi proaktif yang selaras dengan standar yang diakui.
Berpindah ke Electronic Batch Records (EBR) dapat secara signifikan mengurangi kesalahan dokumentasi - hingga 50% - melalui pengumpulan data real-time dan alur kerja otomatis [2]. Sistem ini menandai hasil tes mikroba yang hilang atau ulasan yang tidak lengkap sebelum batch maju, meminimalkan kesalahan manusia.
"FDA mengharapkan catatan untuk menjadi ALCOA(+): Attributable, Legible, Contemporaneous, Original, Accurate - plus Complete, Consistent, Enduring, and Available." – Atlas Compliance [1]
Setiap perbedaan atau penyimpangan yang tidak dapat dijelaskan dalam catatan batch harus dikaitkan dengan investigasi formal dan sistem CAPA. Batasi izin menulis dan menghapus untuk melindungi integritas data uji mikrobiologi elektronik. Produsen kompetitif bertujuan untuk meninjau dan merilis 95% batch dalam waktu 30 hari setelah penyelesaian produksi [2].
Tindakan ini tidak hanya mengurangi risiko kutipan tetapi juga sejalan dengan standar dokumentasi ketat yang disorot dalam inspeksi FDA terbaru.
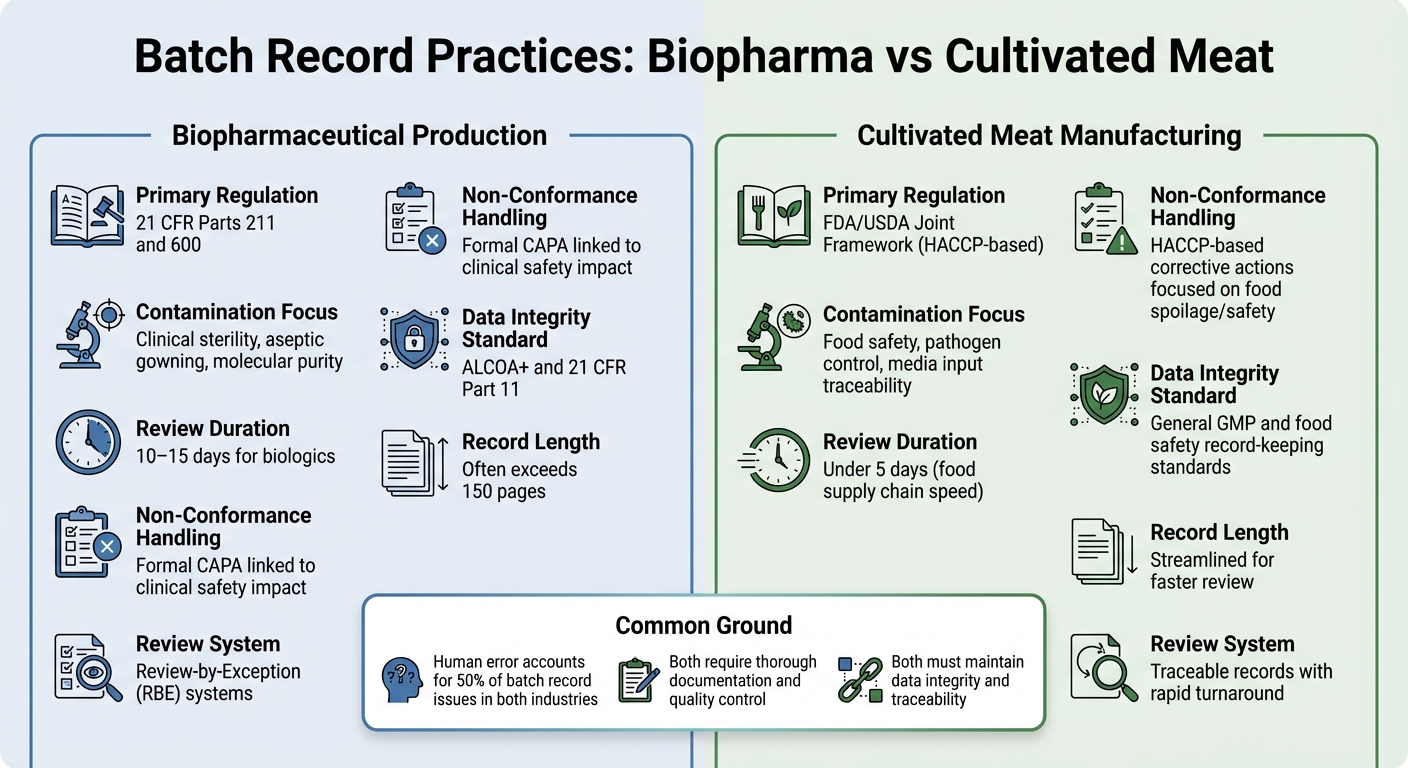
Biopharma vs Daging Budidaya: Perbedaan Catatan Batch
Perbandingan Persyaratan Catatan Batch Biopharmaceutical vs Daging Budidaya
Melihat perbedaan dalam praktik catatan batch antara produksi biopharmaceutical dan manufaktur daging budidaya menawarkan gambaran yang lebih jelas tentang bagaimana tuntutan regulasi membentuk prioritas dokumentasi di industri ini.
Kedua sektor memerlukan dokumentasi yang menyeluruh, tetapi kerangka regulasi dan tujuan pengendalian mereka berbeda secara signifikan. Dalam biopharma, catatan batch diatur ketat di bawah 21 CFR Parts 211 dan 600, yang mengharuskan unit Quality Control untuk meninjau dan menyetujui semua catatan produksi dan pengendalian sebelum batch dapat dirilis [2] . Produsen daging budidaya, di sisi lain, biasanya mengikuti standar HACCP dan GCCP.Ini lebih berfokus pada keamanan pangan dan pengendalian patogen, daripada sterilitas tingkat klinis yang dibutuhkan untuk biologis injeksi.
Catatan batch biopharma sering kali ekstensif, kadang-kadang melebihi 150 halaman, dan proses peninjauan dapat memakan waktu 10–15 hari. Untuk menyederhanakan ini, banyak perusahaan biopharma menggunakan Review-by-Exception (RBE) sistem, yang merangkum penyimpangan kunci menjadi laporan ringkas. Sementara itu, produsen daging budidaya bertujuan untuk catatan yang dapat dilacak yang dapat ditinjau dalam waktu kurang dari lima hari, mencerminkan kecepatan rantai pasokan makanan yang lebih cepat [2] .
Isi dari catatan ini juga menyoroti prioritas yang berbeda. Inspeksi biopharma sering kali berfokus pada detail pemrosesan aseptik, seperti prosedur berpakaian dan kontrol lingkungan. Sebaliknya, catatan daging budidaya harus menekankan input media dan pengujian mikrobiologi untuk memastikan keamanan pangan.Untuk daging budidaya, tantangannya terletak pada pelacakan input media yang kompleks, mendokumentasikan uji mikrobiologi untuk semua bahan, dan memenuhi batas kritis keamanan pangan - tanpa mematuhi persyaratan kemandulan yang lebih ketat dari farmasi.
Tren Kontaminasi dan Ketidaksesuaian
| Fitur | Produksi Biofarmasi | Manufaktur Daging Budidaya |
|---|---|---|
| Regulasi Utama | 21 CFR Bagian 211 dan 600 [2] | Kerangka Kerja Bersama FDA/USDA (berbasis HACCP) |
| Fokus Kontaminasi | Sterilitas klinis, pakaian aseptik, kemurnian molekuler [2] | Keamanan pangan, pengendalian patogen, keterlacakan input media |
| Durasi Tinjauan | 10–15 hari untuk biologis [2] | Di bawah 5 hari (kecepatan rantai pasokan makanan) |
| Penanganan Ketidaksesuaian | CAPA formal terkait dampak keselamatan klinis [2] | Tindakan korektif berbasis HACCP berfokus pada pembusukan/keamanan makanan |
| Standar Integritas Data | ALCOA+ dan 21 CFR Bagian 11 [1] | Standar pencatatan GMP umum dan keamanan makanan |
Sementara tingkat kesalahan manusia serupa di kedua industri - sekitar 50% masalah catatan batch muncul dari kesalahan manusia [2] - taruhannya berbeda.Dalam biopharma, bahkan satu penyimpangan yang tidak terdokumentasi dapat memiliki implikasi serius terhadap keselamatan pasien. Untuk daging yang dibudidayakan, risiko kontaminasi lebih berkaitan dengan patogen bawaan makanan dan pembusukan, yang dapat mempengaruhi seluruh produksi. Kesimpulan Catatan batch berfungsi sebagai log resmi untuk setiap produksi daging yang dibudidayakan - jika suatu langkah tidak dicatat, regulator menganggapnya tidak dilakukan. Ini menyoroti pentingnya dokumentasi yang tepat dan kontrol kualitas yang ketat. Inspeksi FDA menekankan bahwa integritas data harus sesuai dengan prinsip ALCOA+. Tim Pengendalian Kualitas diwajibkan untuk meninjau dan menyetujui semua catatan produksi sebelum suatu batch dapat dirilis [2][17], dan setiap penyimpangan harus segera diselidiki dengan analisis akar penyebab yang terdokumentasi [2] [5]. Sementara kesalahan manusia menyumbang 50% dari masalah catatan batch, tinjauan dua tingkat dan proses CAPA (Tindakan Korektif dan Pencegahan) yang terstruktur dapat membantu mengurangi risiko ini [2][5].
"Bukan kompleksitas proses yang memicu kutipan - melainkan ketidakkonsistenan, ketidaklengkapan, dan pengawasan yang buruk." - GXP Auditing & Layanan Konsultasi [5]
Untuk mengatasi tantangan ini, produsen daging budidaya harus fokus pada audit independen, pengujian ketat bahan makanan yang aman untuk kontaminasi mikrobiologis, dan memastikan dokumentasi mematuhi standar HACCP dan GCCP. Menerapkan sistem catatan batch elektronik, divalidasi di bawah 21 CFR Bagian 11 [1], dapat secara signifikan meminimalkan kesalahan dan mempercepat proses peninjauan.
Lingkungan regulasi menuntut ketelitian, tetapi dapat dinavigasi.Dengan belajar dari kesalahan biopharma - seperti tanda tangan yang hilang di Qinhuangdao Zizhu Pharmaceutical [17], verifikasi ganda yang tidak memadai di Terumo Corp [18], dan dokumentasi penyimpangan yang tidak memadai di Torrent Pharmaceuticals [18] - perusahaan daging budidaya dapat membangun sistem yang sesuai sejak awal. Mengintegrasikan pelajaran ini memungkinkan kepatuhan proaktif dan kualitas yang konsisten. Penyimpanan catatan yang aman, pelaporan penyimpangan tepat waktu, dan melakukan audit tiruan yang realistis akan memastikan catatan batch tetap siap untuk inspeksi dan produksi berjalan sepenuhnya dapat dilacak.
Untuk sumber daya lebih lanjut dan panduan ahli tentang mempertahankan standar produksi tinggi dalam manufaktur daging budidaya, kunjungi
FAQ
Apa yang harus disertakan dalam catatan batch untuk daging hasil budidaya?
Catatan batch untuk daging hasil budidaya berfungsi sebagai log komprehensif dari seluruh proses manufaktur. Ini harus mencakup instruksi pemrosesan terperinci , catatan pelaksanaan langkah demi langkah, dan mencatat setiap penyimpangan yang terjadi selama produksi. Selain itu, harus mendokumentasikan pengujian dalam proses dan pengujian pelepasan untuk memastikan produk memenuhi standar keselamatan, kualitas, dan regulasi.
Bagaimana kita dapat membuktikan kemandulan menggunakan catatan batch?
Membuktikan kemandulan melalui catatan batch melibatkan pemeriksaan menyeluruh terhadap prosedur sterilisasi yang didokumentasikan, hasil pengujian, dan laporan kontrol kualitas media untuk memastikan mereka memenuhi persyaratan regulasi.Sangat penting untuk menangani setiap penyimpangan atau kegagalan tes melalui investigasi mendetail dan CAPA (Tindakan Korektif dan Pencegahan). Proses ini memastikan bahwa setiap langkah telah dipatuhi dan setiap masalah telah diselesaikan dengan benar untuk menjaga standar kesterilan.
Kapan catatan batch elektronik (Bagian 11) diperlukan?
Catatan batch elektronik sangat penting di bawah Bagian 11 ketika sistem elektronik digunakan untuk mendokumentasikan, menyelidiki, dan membenarkan penyimpangan catatan batch. Mereka memainkan peran penting dalam memastikan kepatuhan dengan 21 CFR Part 211.192, menjaga integritas data, memenuhi batas waktu investigasi, dan memastikan pengawasan manajemen yang efektif.









