

배치 기록은 규정 준수 및 제품 안전에 필수적입니다. 이들은 생산의 모든 단계를 문서화하여 규제 기준이 충족되도록 합니다. 배양육 생산자에게는 무균 상태 유지와 상세한 기록이 필수적입니다. FDA 검사는 종종 누락된 데이터, 불완전한 검토, 부적절한 시정 조치와 같은 문제를 강조하며, 이는 경고나 중단으로 이어질 수 있습니다.
핵심 요약:
- 배치 기록: 두 가지 유형 - 마스터 배치 기록(MBR) ("레시피") 및 배치 생산 기록(BPR) ("실행").
- 일반적인 문제: 인간 오류 (문제의 50%), 누락된 공정 중 검사, 불완전한 검토, 부적절한 CAPA (시정 및 예방 조치) 시스템.
- FDA 기준: ALCOA+ 원칙 (속성, 읽기 쉬움, 동시성, 원본, 정확성, 완전성, 일관성, 지속성, 가용성) 준수가 필수입니다.
- 솔루션: 독립적인 감사, 전자 배치 기록, 엄격한 공급업체 검증은 오류를 최소화하고 규정을 준수하는 데 도움이 될 수 있습니다.
UPSIDE Foods와 같은 배양육 회사는 상세한 문서화, 입력 추적 가능성, 신속한 시정 조치를 보장하여 기준을 설정했습니다. 이러한 관행을 배우면 생산자는 규제상의 함정을 피하고 높은 품질 기준을 유지할 수 있습니다.
생명 과학 분야에서 FDA 준수를 위한 문서화 및 기록 보관에 대한 종합 가이드
sbb-itb-ffee270
배치 기록 문서화의 일반적인 문제
FDA 검사 보고서는 일관되게 반복되는 문제를 강조합니다: 생산 기록 검토 편차는 규제 기관이 지적한 주요 GMP 결함 중 하나로 꼽힙니다 [7]. 배양육 생산자에게 이러한 결함은 단순한 행정적 오류를 넘어 지속적인 무균 상태를 입증할 수 있는 능력을 위협합니다. 이러한 문제는 아래 예시에서 설명된 것처럼 여러 형태로 나타납니다.
불완전한 검토 및 비준수
일반적인 문제 중 하나는 품질 관리 부서가 배치 기록을 철저히 검토하지 않는 것입니다. 검토는 출시 과정의 필수적인 부분이 되기보다는 종종 제품 문제가 이미 발생한 후에야 반응적으로 이루어집니다 [7]. 이 접근 방식은 생산 기록에 상당한 공백을 남깁니다.
예를 들어, Davis City Pharmacy는 구성 요소의 양, 운영 단계, 직원의 이니셜과 같은 중요한 세부 사항이 누락된 배치 기록으로 인해 FDA 483 관찰을 받았습니다. 마찬가지로, CAPS 는 주요 항목에 필요한 서명과 검토자 확인이 부족하여 지적되었습니다 [3] . 이러한 실수는 고립된 사건이 아닙니다; 연구에 따르면 약 52%의 문서 위반이 강력한 생물공정 관리 시스템이 없을 때 악화됩니다 [3] .
"프로세스의 복잡성이 인용을 유발하는 것이 아니라 - 일관성 부족, 불완전함, 그리고 부실한 감독입니다." - GXP 감사 & 컨설팅 서비스 [5]
공정 중 검사 기록 누락
또 다른 빈번한 결함은 공정 중 검사에 대한 적절한 문서화의 부재입니다. 이러한 기록은 특히 무균 작업의 중요한 제어 지점에서 중요합니다. 예를 들어, Nephron Sterile Compounding Center는 배치 기록에서 필수적인 가운 착용 단계와 무균 절차를 문서화하지 않아 인용을 받았습니다 [3] . 배양육 생산자에게 있어, 무균 상태가 가장 중요하기 때문에, 이러한 누락은 오염 통제 조치.
의 준수를 확인하는 것을 불가능하게 만듭니다.Amphastar는 또한 예상치 못한 수율 변동이나 출력 불일치를 조사하거나 문서화하지 않은 것으로 지적되었습니다 [3]. 이러한 간과의 위험은 명백합니다. 한 사례에서는, 2024/2025년의 미확인 제약 시설이 완성된 배치 기록을 개방형 선반과 책상에 보관하고 있는 것이 발견되었습니다. FDA 조사관들은 단일 기록에서 7페이지를 포함한 누락된 페이지와 다른 기록에서 "합성 솔루션" 섹션 전체가 없는 것을 발견했습니다 [6].
CAPA 및 공급업체 GMP 검증 실패
문서 오류를 넘어, 효과적인 시정 절차와 공급업체 검증의 부재는 배치 기록의 신뢰성을 더욱 저해합니다.생산 편차가 해당하는 시정 및 예방 조치(CAPA) 보고서 없이 발생하면 배치 기록의 무결성이 손상됩니다 [7]. 예를 들어, Eugia Pharma Specialities Limited, 는 2024년 1월 22일부터 2월 2일까지 검사받았으며, 불일치를 적절히 검토하지 않아 FDA 483을 받았습니다. 그들의 비효율적인 CAPA 시스템과 불완전한 조사는 반복적인 생산 문제를 초래하여 조사 및 CAPA 절차의 전면적인 개편을 강요했습니다 [9].
마찬가지로, 2023년 9월 26일부터 10월 25일까지의 검사 동안, Stokes Healthcare Inc.는 불량한 불일치 관리를 보여주었습니다. 회사는 모든 영향을 받은 배치에 대한 조사를 확장하지 못했고 분석 완료를 지연시켰습니다 [9].
"CAPA 또는 편차 보고서가 없는 불일치? 그것은 규정 준수 실패입니다." - GXP 감사 & 컨설팅 서비스 [5]
공급업체 검증 문제는 또 다른 복잡성을 추가합니다. Empower Clinic Services LLC는 2022년 7월 18일부터 8월 5일까지의 검사에서 불충분한 공급업체 자격 및 부실한 조사 절차를 포함한 부적절한 품질 관리 절차로 지적되었습니다 [9] . 성장 배지, 세포주 및 기타 중요한 투입물에 의존하는 배양육 생산자에게는 공급업체의 GMP 준수가 배치 기록의 무결성을 유지하는 데 필수적입니다.
배치 기록에 대한 FDA 요구 사항
배치 기록에 대한 FDA의 규칙은 21 CFR Part 117, 을 중심으로 하며, 이는 식품 안전의 기준을 설정합니다.배양육의 경우, 세포 배양 단계에서 무균 상태를 유지하는 것이 중요하며, 문서는 종종 Part 111 또는 Part 211 , 의 더 엄격한 기준을 충족해야 합니다. Part 117 [10][14]. 이것은 배양육 생산의 안전성과 효과성을 보장하기 위해 정밀한 문서화가 얼마나 중요한지를 강조합니다.
배치 기록의 핵심 표준
각 배치에는 두 가지 주요 문서가 필요합니다:
BPR에는 배치 또는 로트 번호, 장비 세부 정보, 청소 날짜, 구성 요소 식별자, 정확한 측정값, 실제 수율과 이론적 수율의 비교와 같은 세부 사항이 포함되어야 합니다 [10][14].
"배치 생산 기록은 적절한 마스터 제조 기록을 정확하게 따라야 하며, 배치 생산의 각 단계를 수행해야 합니다." – 21 CFR 111.255 [12]
모든 중요한 단계는 즉시 기록되어야 하며, 수행자와 검증자의 이니셜이 기재되어야 합니다 [10][11]. FDA는 ALCOA(+) 원칙을 준수할 것을 요구하며, 이는 기록이 속성 가능하고, 읽을 수 있으며, 동시적이고, 원본이며, 정확해야 함을 의미합니다 - 또한 완전하고, 일관되며, 지속 가능하고, 이용 가능해야 합니다 [1] .
마스터 제조 기록에서 어떤 편차가 발생할 경우, 철저히 조사해야 합니다. 여기에는 문제 문서화, 근본 원인 분석 수행, 시정 및 예방 조치 (CAPA) 계획 실행이 포함됩니다 [8] [1]. 편차의 초기 평가는 감지 후 24–48시간 이내에 기록되어야 합니다 [8]. 전자 시스템을 사용하는 시설의 경우, 21 CFR Part 11 준수가 필수적입니다. 여기에는 검증된 전자 서명과 안전하고 시간 스탬프가 찍힌 감사 추적이 포함됩니다 [8] [1].
기록 보존 및 검토 절차
적절한 기록 보존 및 검토 절차는 규정 준수 유지와 제품 안전 보장을 위해 중요합니다.멸균 생산, 예를 들어 배양육의 경우, 배치 기록의 모든 세부 사항은 철저한 검토를 거쳐야 합니다. 품질 관리 (QC) 팀은 배치가 유통 승인을 받기 전에 모든 배치 기록을 검토하고, 결과를 모니터링하며, 테스트 데이터를 확인하는 책임을 집니다 [10] [13].
"모든 의약품 생산 및 관리 기록은 배치가 출시되거나 유통되기 전에 품질 관리 부서에 의해 검토되고 승인되어야 합니다." – 21 CFR 211.192 [2]
제조업체는 종종 생산 후 30일 이내에 배치 검토의 95%를 완료하는 것을 목표로 합니다 [2] . 그러나 배양육에 관련된 더 복잡한 멸균 공정의 경우, 검토는 일반적으로 7–10일, 이 소요되며, 성능이 우수한 시설은 7일 이내에 완료합니다 [2]. 전자 배치 기록 시스템은 배양육 생산 시스템, 에 통합된 경우와 같이 검토 속도를 크게 높일 수 있습니다. 종이 기반 방법에 비해 시간이 절반으로 줄어들며, Part 11 요구 사항을 충족하고 데이터 무결성을 유지하도록 검증된 경우에만 가능합니다.[1].
FDA 승인 배양육 회사들이 성공한 이유
FDA 승인 배양육 회사들은 문서화 문제를 해결하고 엄격한 안전 기준을 충족하는 관행을 채택하여 높은 기준을 설정했습니다.
UPSIDE Foods가 2022년 11월에 FDA의 사전 시장 상담을 통과한 최초의 배양육 회사가 되었을 때, 그들은 업계를 위한 모델을 수립했습니다.FDA는 세포주 확립, 세포 은행, 제조 관리 및 모든 구성 요소와 입력을 포함한 생산 과정을 철저히 검토한 후 "추가 질문 없음"이라는 서신을 발행했습니다.[16]. 이 성과는 FDA의 엄격한 요구 사항을 충족하기 위한 상세한 문서화의 중요성을 강조했습니다.
무균 및 준수 기준 충족
UPSIDE Foods의 두드러진 성과는 입력 추적 가능성에 대한 철저한 접근 방식이었습니다. 모든 생산 구성 요소가 신중하게 문서화되어 초기 세포주부터 최종 제품까지 명확한 책임 체인이 보장되었습니다.[16]. 이러한 수준의 투명성 덕분에 FDA 검토자들은 제조 과정의 모든 단계를 추적할 수 있었으며, 모든 안전 기준이 일관되게 충족되었음을 확인할 수 있었습니다.
"FDA의 사전 시장 협의에는 회사의 생산 공정과 생산 공정에 의해 만들어진 배양 세포 물질의 평가가 포함되었습니다. 여기에는 1차 및 불멸화 세포주와 세포 은행, 제조 관리, 모든 구성 요소 및 입력의 설정이 포함됩니다." – U.S. 식품의약국 [16]
다른 성공적인 회사들은 세부적인 무균 공정 문서를 구현하여 뒤따랐습니다. 여기에는 가운 착용 절차 및 무균 취급 작업과 같은 중요한 단계가 포함되었습니다 [3]. 이전의 문서화 실패와 달리, 이러한 회사들은 운영자 확인, 생산 감독, 품질 부서 검토를 포함하는 계층화된 검토 시스템을 사용하여 배치 출시 전에 잠재적인 오류를 잡았습니다 [15]. 전자 배치 기록 시스템은 각 단계에서 필수 서명을 강제하고 21 CFR Part 11 요구 사항에 따라 변경 불가능한 감사 추적을 유지하는 데 중요한 역할을 했습니다 [3][2].
이러한 엄격한 관행은 자연스럽게 회사가 편차와 실패를 처리하는 방식으로 확장되었습니다.
배치 실패에 대한 CAPA 프로세스
배치가 사양을 충족하지 못했을 때, FDA 승인 회사는 신속하고 체계적인 조치를 취했습니다. 그들의 시정 및 예방 조치(CAPA) 프로세스에는 공식적인 근본 원인 분석, 영향 평가 및 명확하게 문서화된 시정 조치가 포함되었습니다 [3]. 모든 편차는 통합 품질 보증 프레임워크 내에서 관리되어 모든 문제가 철저히 조사되고 정당화되며 문서화된 후에 생산이 계속되도록 보장했습니다 [2].
앞으로 데이터 무결성은 2024-2025년 동안 FDA 집행 조치의 주요 초점이 될 것입니다 [1].
배치 기록 관행을 개선하는 방법
배치 기록 관행을 강화하려면 FDA 검사 중에 자주 식별되는 일반적인 실패를 해결하기 위해 정확한 문서화가 필요합니다. 주요 과제를 해결하기 위한 몇 가지 전략이 있습니다.
독립적인 배치 기록 감사 수행
정기적인 제3자 감사는 내부 검토에서 간과할 수 있는 문제를 발견할 수 있습니다. 실험실 정보 관리 시스템(LIMS), 제조 실행 시스템(MES), 전사적 자원 관리(ERP)와 같은 중요한 시스템에 초점을 맞추는 것부터 시작하십시오. 방출 시험 기록, 안정성 데이터, 배치 생산 기록과 같은 규제 영향이 큰 문서를 우선시하십시오.
효과적인 방법 중 하나는 샘플 회수 테스트입니다.무작위로 최근 배치를 선택하고 생산 및 실험실 기록을 재구성하십시오. 이는 누락된 데이터, 불완전한 서명 또는 문서화 격차를 찾아내어 규제 위반을 초래할 수 있는 문제를 식별하는 데 도움이 될 수 있습니다. 시스템 생성 감사 추적을 수동 입력과 교차 확인하여 승인되지 않은 변경 또는 삭제를 식별하십시오.
지난 1년간의 Out-of-Specification (OOS) 및 Out-of-Trend (OOT) 보고서를 검토하십시오. 근본 원인 분석이 철저했는지, 시정 및 예방 조치(CAPA)가 적절히 구현되었는지 평가하십시오. 문서화 문제는 FDA 경고 서신의 21%를 차지하며, 인적 오류는 제약 제조에서 배치 기록 문제의 50%를 차지한다는 점을 주목할 가치가 있습니다 [2].
"프로세스의 복잡성이 아니라 일관성 부족, 불완전함, 부실한 감독이 위반을 유발합니다." – GXP 감사 & 컨설팅 서비스 [5]
주기적인 모의 검토를 통해 규제 검사를 시뮬레이션합니다. 이 실천은 팀이 실제 감사 전에 불일치 및 잠재적인 데이터 무결성 문제를 인식하는 데 도움이 됩니다. 모든 기록이 ALCOA+ 원칙을 따르는지 확인하십시오: 속성, 가독성, 동시성, 원본, 정확성, 완전성, 일관성, 지속성, 가용성.
문서 무결성이 확고해지면 모든 생산 입력의 품질을 검증하는 데 집중하십시오.
모든 입력의 미생물 오염 테스트
모든 입력에 대한 독립적인 무균 및 효능 테스트는 필수적입니다 - 공급업체의 분석 증명서(CoAs)에만 의존하지 마십시오. 이는 특히 배양육 생산자에게 중요하며, 오염은 전체 배치를 위태롭게 할 수 있습니다.
예를 들어, 2013년 2월에 Central Admixture Pharmacy Services는 무균 제품의 배치 출시 중 미생물 제어가 불충분하여 FDA의 지적을 받았습니다. 회사는 표준 운영 절차(SOP)에 상세한 미생물 제어 절차를 도입해야 했습니다.[4].
공정 중 미생물 체크포인트는 최종 제품 테스트에 대한 과도한 의존을 방지할 수 있습니다. 이러한 체크포인트를 배치 출시 SOP에 통합하고 엄격한 동시 문서를 유지하십시오. 모든 테스트 결과와 제조 단계를 기록하여 백데이트나 지연된 입력을 피하고, 이는 FDA의 지적을 초래할 수 있습니다.
CoA, 감사 보고서, 품질 계약 및 입고 자재와 관련된 모든 편차의 이력을 포함하여 포괄적인 공급업체 파일을 유지하십시오.
배치 기록 관행을 강화하는 것은 HACCP 및 GCCP와 같은 확립된 표준과 프로세스를 정렬하는 것을 포함합니다.
HACCP 및 GCCP 표준에 맞춘 기록 정렬
위해요소분석 중요관리점(HACCP) 원칙을 배치 기록에 통합하면 생산 전반에 걸쳐 중요한 공정 변수가 모니터링되고 문서화됩니다. 이는 최종 단계 테스트에만 의존하는 대신 공정 중 미생물 테스트 체크포인트를 설정하는 것을 포함합니다.
배양육 생산자에게는 우수 세포 배양 실습(GCCP) 표준 준수가 동일하게 중요합니다. 배치 기록에는 무균 조작, 가운 착용 절차 및 배치 출시 기준과 관련된 환경 모니터링 세부 정보가 포함되어야 합니다 [3][4]. 이러한 단계는 규정 준수를 유지하고 제품 안전을 보장하는 데 도움이 됩니다.
산업 데이터에 따르면 적절한 배치 제조 소프트웨어가 없을 경우 문서 위반의 52%가 증가합니다 [3][4]. 사례로, 2023년 2월에 Nephron Sterile Compounding Centre는 배치 출시 전에 중요한 프로세스 변수를 검증하기 위한 제어 절차의 부재로 인해 FDA의 관찰을 받았습니다 [4]. 이는 인정된 표준에 맞춘 사전 문서화의 필요성을 강조합니다.
전자 배치 기록(EBR)으로 전환하면 실시간 데이터 수집과 자동화된 워크플로를 통해 문서 오류를 최대 50%까지 크게 줄일 수 있습니다 [2]. 이 시스템은 배치가 진행되기 전에 누락된 미생물 테스트 결과나 불완전한 검토를 표시하여 인적 오류를 최소화합니다.
"FDA는 기록이 ALCOA(+)이어야 한다고 기대합니다: 속성, 읽기 쉬움, 동시성, 원본, 정확성 - 그리고 완전성, 일관성, 지속성, 가용성을 포함합니다." – Atlas Compliance [1]
배치 기록에서 설명되지 않은 불일치 또는 편차는 공식 조사 및 CAPA 시스템과 연결되어야 합니다. 전자 미생물학적 시험 데이터의 무결성을 보호하기 위해 쓰기 및 삭제 권한을 제한하십시오. 경쟁 제조업체는 생산 완료 후 30일 이내에 배치의 95%를 검토하고 출시하는 것을 목표로 합니다 [2].
이러한 조치는 인용 위험을 줄일 뿐만 아니라 최근 FDA 검사에서 강조된 엄격한 문서화 표준과도 일치합니다.
바이오제약 vs 배양육: 배치 기록 차이점
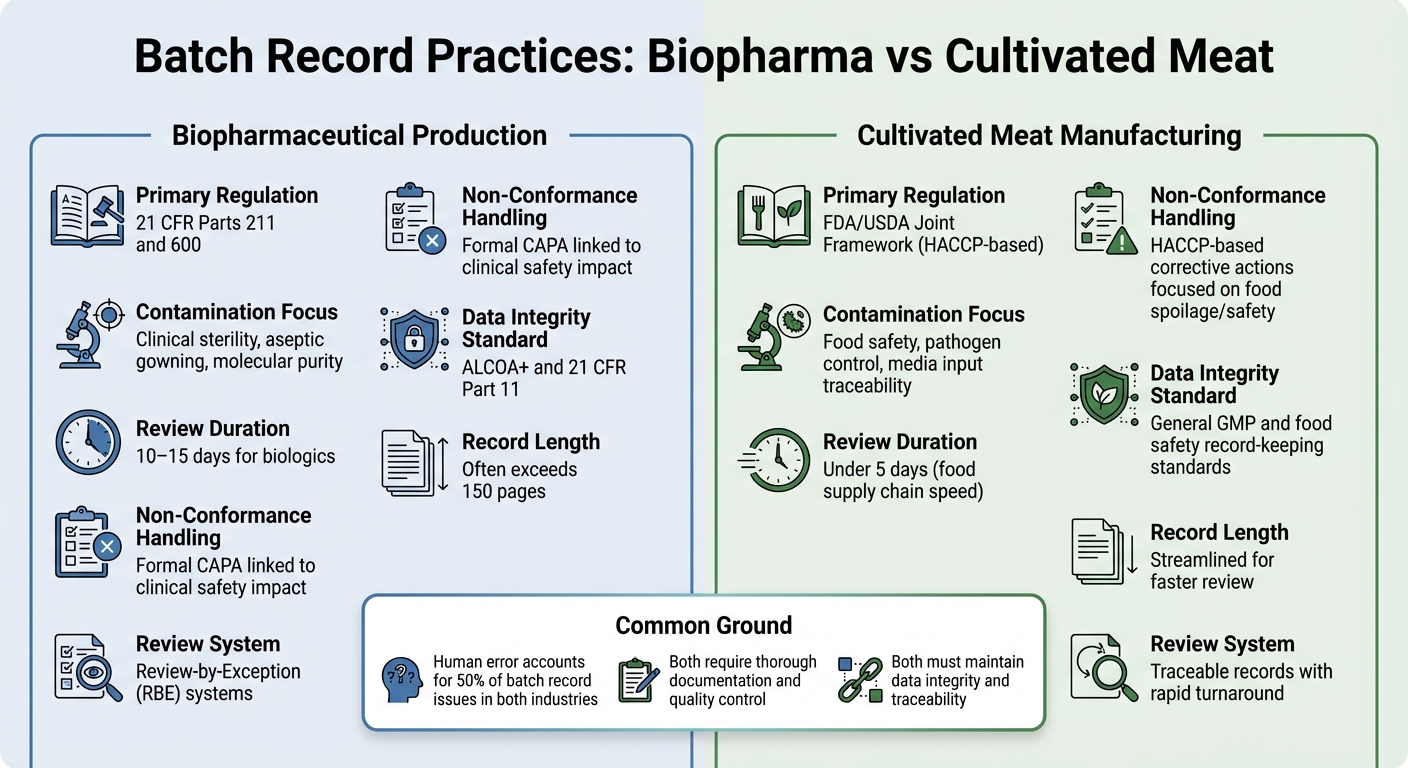
바이오제약 vs 배양육 배치 기록 요구사항 비교
바이오제약 생산과 배양육 제조의 배치 기록 관행의 차이를 살펴보면, 이러한 산업에서 규제 요구가 문서화 우선순위에 어떻게 영향을 미치는지 더 명확하게 알 수 있습니다.
두 분야 모두 철저한 문서화가 필요하지만, 그들의 규제 프레임워크와 통제 목표는 상당히 다릅니다. 바이오제약에서는 배치 기록이 21 CFR Parts 211 and 600, 에 따라 엄격하게 규제되며, 품질 관리 부서가 배치가 출시되기 전에 모든 생산 및 통제 기록을 검토하고 승인해야 합니다 [2]. 반면에 배양육 생산자는 일반적으로 HACCP 및 GCCP 표준을 따릅니다.이들은 주사 가능한 생물학 제제에 요구되는 임상 등급의 무균성보다는 식품 안전과 병원체 제어에 더 중점을 둡니다.
바이오제약 배치 기록은 종종 방대하며, 때로는 150페이지를 초과하기도 하고 검토 과정은 10~15일이 걸릴 수 있습니다. 이를 간소화하기 위해 많은 바이오제약 회사들은 예외에 의한 검토 (RBE) 시스템을 사용하여 주요 편차를 요약한 간결한 보고서를 작성합니다. 한편, 배양육 생산업체들은 식품 공급망의 빠른 속도를 반영하여 5일 이내에 검토할 수 있는 추적 가능한 기록을 목표로 합니다 [2].
이 기록의 내용은 또한 우선순위의 차이를 강조합니다. 바이오제약 검사는 종종 가운 착용 절차 및 환경 통제와 같은 무균 처리 세부 사항에 중점을 둡니다. 반면, 배양육 기록은 식품 안전을 보장하기 위해 배지 투입물 및 미생물학적 테스트를 강조해야 합니다.배양육의 경우, 복잡한 배지 입력을 추적하고 모든 재료에 대한 미생물학적 테스트를 문서화하며, 제약의 엄격한 무균 요구 사항을 준수하지 않으면서 식품 안전 중요 한계를 충족하는 것이 과제입니다.
오염 및 비준수 동향
| 특징 | 바이오의약품 생산 | 배양육 제조 |
|---|---|---|
| 주요 규제 | 21 CFR 파트 211 및 600[2] | FDA/USDA 공동 프레임워크 (HACCP 기반) |
| 오염 집중 | 임상 무균성, 무균 가운 착용, 분자 순도[2] | 식품 안전, 병원체 제어, 매체 입력 추적 가능성 |
| 검토 기간 | 생물학적 제제의 경우 10-15일[2] | 5일 이내 (식품 공급망 속도) |
| 비준수 처리 | 임상 안전 영향과 관련된 공식 CAPA [2] | 식품 부패/안전에 중점을 둔 HACCP 기반 시정 조치 |
| 데이터 무결성 표준 | ALCOA+ 및 21 CFR Part 11 [1] | 일반 GMP 및 식품 안전 기록 보관 표준 |
두 산업 모두에서 인간 오류율은 비슷하지만 - 배치 기록 문제의 약 50%는 인간의 실수에서 발생합니다 [2] - 위험은 다릅니다.바이오 제약에서는 단 하나의 문서화되지 않은 편차도 환자 안전에 심각한 영향을 미칠 수 있습니다. 배양육의 경우, 오염 위험은 식중독 병원균과 부패에 더 관련이 있으며, 이는 전체 생산 과정에 영향을 미칠 수 있습니다.
결론
배치 기록은 모든 배양육 생산 과정의 공식 기록으로 사용됩니다 - 만약 어떤 단계가 기록되지 않으면, 규제 기관은 그것이 수행되지 않은 것으로 간주합니다 [6] [3]. 이는 정확한 문서화와 엄격한 품질 관리의 중요성을 강조합니다.
FDA 검사는 데이터 무결성이 ALCOA+ 원칙과 일치해야 한다고 강조합니다 [1]. 품질 관리 팀은 배치가 출시되기 전에 모든 생산 기록을 검토하고 승인해야 합니다 [2][17], 그리고 모든 편차는 문서화된 근본 원인 분석과 함께 신속하게 조사되어야 합니다 [2][5]. 인적 오류가 배치 기록 문제의 50%를 차지하지만, 이중 수준의 검토와 구조화된 CAPA(시정 및 예방 조치) 프로세스는 이러한 위험을 줄이는 데 도움이 될 수 있습니다 [2][5].
"프로세스의 복잡성이 인용을 유발하는 것이 아니라 일관성 부족, 불완전함, 그리고 부실한 감독입니다." - GXP 감사 & 컨설팅 서비스 [5]
이러한 도전을 극복하기 위해, 배양육 생산업체는 독립적인 감사, 미생물 오염에 대한 식품 안전 성분의 철저한 테스트, 그리고 문서가 HACCP 및 GCCP 표준을 준수하도록 보장하는 데 집중해야 합니다. 21 CFR Part 11에 따라 검증된 전자 배치 기록 시스템을 구현하면 오류를 크게 줄이고 검토 과정을 가속화할 수 있습니다.
규제 환경은 정밀성을 요구하지만, 탐색 가능합니다.Qinhuangdao Zizhu Pharmaceutical [17] , Terumo Corp[18], Torrent Pharmaceuticals [18] - 바이오 제약의 오류, 예를 들어 서명 누락, 이중 검증 부족, 불충분한 편차 문서화 등을 통해 배양육 회사들은 처음부터 준수 시스템을 구축할 수 있습니다. 이러한 교훈을 통합하면 사전 준수 및 일관된 품질을 보장할 수 있습니다. 안전한 기록 보관, 적시 편차 보고, 현실적인 모의 감사 실시를 통해 배치 기록이 검사 준비 상태를 유지하고 생산 실행이 완전히 추적 가능하도록 할 것입니다.
배양육 제조에서 높은 생산 기준을 유지하기 위한 추가 자료 및 전문가 지침을 보려면
자주 묻는 질문
배양육의 배치 기록에는 무엇이 포함되어야 하나요?
배양육의 배치 기록은 전체 제조 과정을 포괄적으로 기록하는 로그입니다. 여기에는 상세한 처리 지침, 단계별 실행 기록, 및 생산 중 발생하는 이탈 사항을 기록해야 합니다. 또한, 제품이 안전성, 품질 및 규제 기준을 충족하는지 확인하기 위해 공정 중 시험 및 출하 시험을 문서화해야 합니다.
배치 기록을 통해 무균성을 어떻게 증명할 수 있나요?
배치 기록을 통한 무균성 증명은 문서화된 멸균 절차, 시험 결과 및 매체 품질 관리 보고서를 철저히 검토하여 규제 요구 사항을 충족하는지 확인하는 것을 포함합니다.모든 편차나 실패한 테스트는 상세한 조사와 CAPA (시정 및 예방 조치)를 통해 해결하는 것이 중요합니다. 이 과정은 모든 단계가 준수되었고, 문제가 적절히 해결되어 무균 기준을 유지하도록 보장합니다.
전자 배치 기록(Part 11)은 언제 필요합니까?
전자 배치 기록은 전자 시스템이 배치 기록 편차를 문서화, 조사 및 정당화하는 데 사용될 때 Part 11에 따라 필수적입니다. 이는 21 CFR Part 211.192, 의 준수를 보장하고, 데이터 무결성을 보호하며, 조사 기한을 충족하고, 효과적인 관리 감독을 보장하는 데 중요한 역할을 합니다.









