

Em salas limpas, conformidade com GMP garante qualidade e segurança consistentes ao exigir monitoramento detalhado e registros de dados precisos. Para instalações de carne cultivada, isso é particularmente importante, pois até mesmo pequenas variações nas condições da sala limpa podem comprometer o crescimento celular ou contaminar lotes de produção.
Pontos principais:
- Padrões GMP: Foco na integridade dos dados, seguindo o framework ALCOA+ (Atribuível, Legível, Contemporâneo, Original, Preciso, Completo, Consistente, Duradouro, Disponível).
- Parâmetros Críticos: Monitore partículas de ar, contagens microbianas, temperatura, umidade e pressão para detectar riscos precocemente. Isso requer selecionar sensores precisos capazes de manter esses parâmetros críticos.
- Sistemas de Dados: Use sistemas de controle de bioprocessos validados com acesso baseado em funções, trilhas de auditoria e armazenamento seguro para registros eletrônicos e em papel.
- Riscos Comuns: Evite erros no manuseio manual de dados, alterações de configuração sem controle e práticas inadequadas de armazenamento.
- GMP Personalizado para Carne Cultivada: Ajuste as estratégias de monitoramento para abordar riscos únicos, como condições de biorreatores e resíduos de agentes de limpeza.
Para P&D de carne cultivada, o gerenciamento robusto de dados garante a segurança do produto, conformidade regulatória e operações escaláveis. Aborde vulnerabilidades conhecidas de forma proativa para evitar problemas regulatórios custosos no futuro.
Requisitos Chave de GMP para Integridade de Dados em Salas Limpas
Compreendendo os Princípios ALCOA+
A pedra angular da integridade de dados GMP reside no ALCOA+ framework. Órgãos reguladores como o MHRA , EMA, e OMS o utilizam para determinar se os registros de salas limpas podem ser confiáveis.ALCOA+ significa: Atribuível, Legível, Contemporâneo, Original, Preciso, Completo, Consistente, Duradouro, e Disponível . Cada um desses termos tem importância prática nas operações de sala limpa.
- Atribuível: Cada entrada - seja uma contagem de partículas, leitura de pressão ou registro de limpeza - deve mostrar claramente quem a registrou, junto com a data, hora e detalhes relevantes do instrumento.
- Legível: Os registros devem ser fáceis de ler e decifrar, garantindo clareza durante revisões ou inspeções.
- Contemporâneo: Os dados devem ser registrados em tempo real. Entradas atrasadas ou retrospectivas podem comprometer a confiabilidade dos registros.
- Original: Os dados devem permanecer em sua forma capturada inicialmente, sem edições ou alterações não autorizadas.
- Preciso: Os valores registrados devem refletir verdadeiramente os resultados observados, livres de erros ou manipulações.
- Completo: Todos os registros relevantes, incluindo desvios ou resultados fora das especificações, devem ser documentados.
- Consistente, Duradouro, e Disponível: Os registros devem seguir a sequência correta, ser preservados intactos pelo período de retenção exigido e estar prontamente acessíveis para revisão ou inspeção.
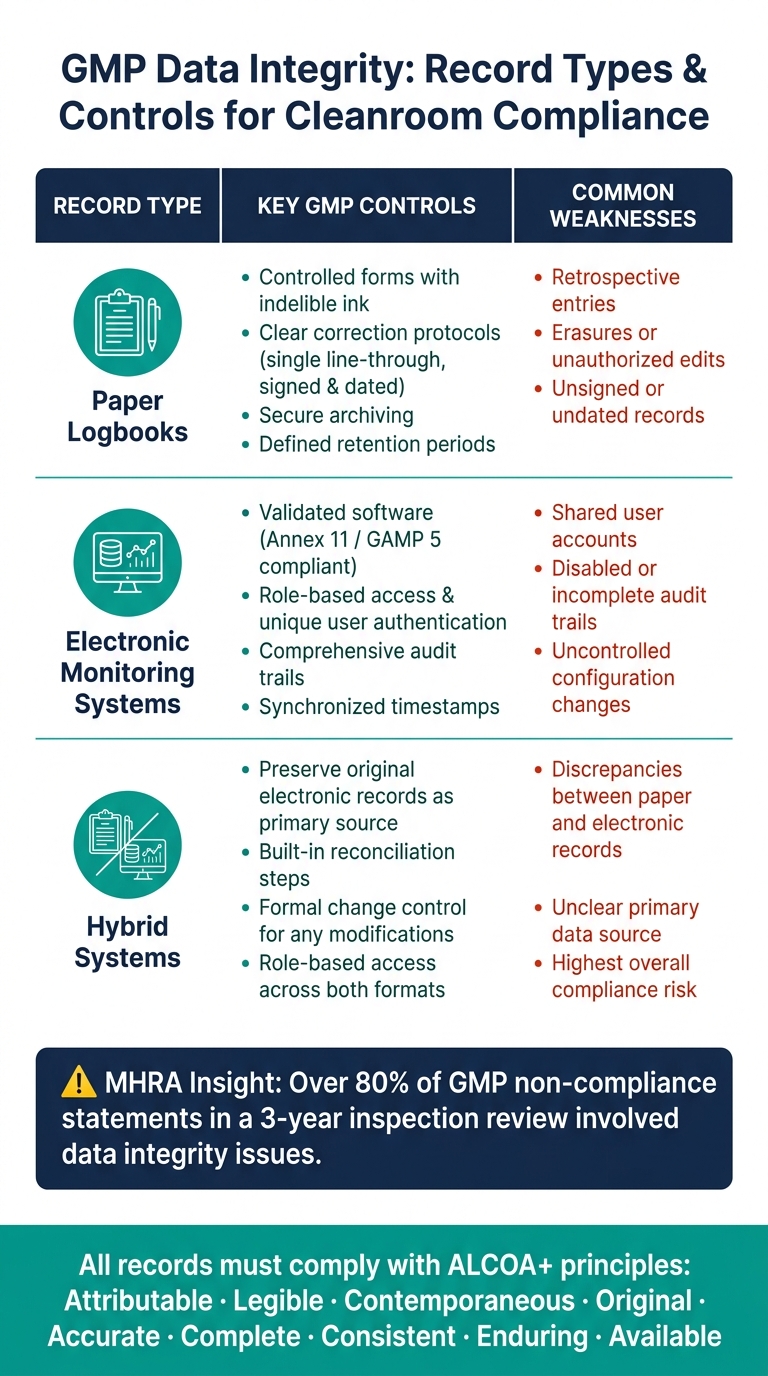
Os reguladores dão grande ênfase a esses princípios. Por exemplo, uma revisão de inspeção da MHRA revelou que mais de 80% das declarações de não conformidade com as BPF em um período de três anos envolveram questões de integridade de dados [5]. Para incorporar o ALCOA+ nos fluxos de trabalho diários, as instalações podem adotar formulários bem estruturados, impor campos obrigatórios e realizar revisões regulares de trilhas de auditoria.
Com ALCOA+ como base, o próximo passo é garantir que esses princípios sejam mantidos em sistemas de papel, eletrônicos e híbridos.
Garantindo a Integridade dos Dados em Diferentes Formatos
Em instalações de carne cultivada, onde os dados influenciam diretamente as decisões de liberação de lotes, manter a integridade em todos os formatos de registro é inegociável. As BPF exigem o mesmo nível de integridade para registros em papel e eletrônicos, embora os controles específicos possam diferir dependendo do formato.
- Sistemas de papel: As melhores práticas incluem o uso de formulários controlados com tinta indelével e protocolos claros de correção (e.g. , correções com uma linha através, assinaturas e datas). Arquivamento seguro e adesão a períodos de retenção definidos também são críticos.
- Sistemas eletrônicos: Estes devem operar em software validado que esteja em conformidade com Annex 11 e GAMP 5. Principais recursos incluem acesso baseado em funções, autenticação de usuário única, trilhas de auditoria abrangentes e carimbos de data/hora sincronizados. Revisões regulares das trilhas de auditoria são essenciais para identificar e resolver quaisquer irregularidades.
- Sistemas híbridos: Estes representam o maior risco, pois envolvem registros eletrônicos e em papel. Por exemplo, quando um instrumento gera dados eletrônicos que são posteriormente transcritos em um registro em papel, a saída eletrônica original deve ser preservada como o registro principal. Etapas de reconciliação devem ser incorporadas ao fluxo de trabalho para detectar e resolver quaisquer discrepâncias entre os registros eletrônicos e em papel. Isso é particularmente crítico na produção de carne cultivada, onde até mesmo pequenas inconsistências de dados podem comprometer medidas de controle de contaminação.
A tabela abaixo resume os controles principais e as fraquezas comuns para cada tipo de registro:
| Tipo de Registro | Controles GMP Principais | Fraquezas Comuns |
|---|---|---|
| Formulários controlados, tinta indelével, protocolos claros de correção, entradas assinadas e datadas | Entradas retrospectivas, rasuras, registros não assinados | |
| Sistemas de monitoramento eletrônico | Software validado, acesso baseado em funções, trilhas de auditoria, sincronização de tempo | Contas de usuário compartilhadas, trilhas de auditoria desativadas ou incompletas |
| Sistemas híbridos | Preservar registros eletrônicos originais; implementar etapas de reconciliação | Discrepâncias entre registros em papel e eletrônicos, fonte de dados primária não clara |
Para garantir a conformidade, os registros devem ser categorizados de acordo com sua criticidade.Para instalações de carne cultivada, dados relacionados a decisões de liberação de lotes ou controle de contaminação (e.g. , resultados de monitoramento ambiental, logs de alarme de HVAC, ou dados de teste de integridade de filtro) devem estar sujeitos aos controles de acesso mais rigorosos, revisões frequentes e gestão robusta de trilhas de auditoria.
sbb-itb-ffee270
Monitoramento Ambiental de Rotina em Sala Limpa GMP & Integridade de Dados 21CFR parte 11
Gerenciamento do Ciclo de Vida dos Dados da Sala Limpa
Integridade de Dados GMP: Tipos de Registro & Controles para Conformidade de Sala Limpa
Estágios do Ciclo de Vida dos Dados da Sala Limpa
O ciclo de vida dos dados da sala limpa em instalações de carne cultivada envolve várias etapas, cada uma com requisitos específicos de conformidade.
Geração de dados marca o início.Isso inclui leituras de instrumentos como contadores de partículas, sensores de pressão diferencial, sondas de temperatura e umidade, amostradores de ar viável, placas de contato de superfície e registros de verificação de limpeza. Para cada parâmetro, deve haver uma frequência de amostragem documentada, um operador designado e um instrumento calibrado. Alinhar essas tarefas de monitoramento com as etapas de produção - como inoculação, expansão celular ou colheita - ajuda a demonstrar como o controle ambiental está diretamente ligado à qualidade e segurança do produto.
Uma vez gerados, os dados entram na fase de captura e transferência . Idealmente, os sistemas eletrônicos devem registrar automaticamente as leituras com entradas carimbadas com data e hora vinculadas a contas de usuários individuais. Para entradas baseadas em papel, os dados devem ser registrados em tempo real usando tinta indelével, com verificações de reconciliação em vigor ao transferir os dados para sistemas eletrônicos.
A fase de armazenamento é igualmente crítica.Tanto os dados brutos quanto os processados devem ser preservados para que qualquer valor relatado possa ser rastreado até seu registro original. Isso requer repositórios seguros e validados com controles de acesso baseados em funções e testes regulares de backup. Os backups devem ser armazenados em um local separado do sistema principal e verificados periodicamente para garantir que possam ser restaurados quando necessário.
Finalmente, o arquivamento conclui o ciclo de vida. Os registros passam para um estado de acesso controlado e somente leitura quando não são mais usados ativamente, mas devem permanecer recuperáveis pelo período de retenção exigido. Em instalações de carne cultivada, o arquivamento de dados da fase de desenvolvimento também pode apoiar esforços futuros de validação.
Uma compreensão clara dessas etapas é essencial para gerenciar riscos de forma eficaz, conforme descrito abaixo.
Riscos Comuns na Gestão de Dados
O manuseio de dados durante transferências apresenta riscos consideráveis.Erros de transcrição manual e entradas retrospectivas podem comprometer a integridade dos dados. Para evitar isso, todas as entradas devem aderir aos princípios ALCOA+ (Atribuível, Legível, Contemporâneo, Original, Preciso, além de Completo, Consistente, Duradouro e Disponível) em tempo real.
Mudanças de configuração são outra preocupação importante. Ajustes nos limites de alarme, mapeamentos de sensores ou configurações do relógio do sistema sem controle formal de mudanças podem comprometer a confiabilidade dos dados registrados antes e depois da mudança. Além disso, falhas de armazenamento - seja devido a bancos de dados corrompidos, backups não testados ou arquivos em papel danificados por fatores ambientais - podem tornar registros críticos inacessíveis. Para mitigar esses riscos, garanta que cada fluxo de dados seja mapeado para um ponto de arquivamento designado com propriedade clara, reduzindo a probabilidade de vulnerabilidades serem identificadas durante inspeções regulatórias.
Controles GMP para Sistemas de Monitoramento de Salas Limpas
Controles Críticos para Sistemas de Monitoramento
Um sistema de monitoramento que atende aos padrões GMP depende de sensores calibrados, manuseio seguro de dados e gerenciamento eficaz de alarmes. Sensores para parâmetros como temperatura, umidade relativa, pressão diferencial, contagem de partículas não viáveis e amostragem microbiana viável devem ser calibrados de acordo com cronogramas documentados e rastreáveis a padrões reconhecidos. Automatizar a transferência de dados desses sensores, completa com carimbos de tempo sincronizados, minimiza o risco de erros manuais.
O gerenciamento de alarmes é igualmente crucial. Os limites de alarme devem estar alinhados com estruturas regulatórias como os limites de classe da ISO 14644-1 e as diretrizes do EU GMP Anexo 1. Cada alarme acionado deve ser acompanhado por uma resposta registrada, incluindo detalhes do usuário, carimbos de tempo e quaisquer comentários.Falhar em documentar uma resposta de alarme cria uma vulnerabilidade de conformidade.
Os controles de acesso baseados em função devem ser rigorosamente aplicados em todo o sistema. A autorização em nível de administrador deve ser exigida para quaisquer alterações nos limites de alarme, configurações de sensores ou configurações do relógio do sistema, e essas alterações devem seguir um processo formal de controle de mudanças. Trilhas de auditoria são obrigatórias para todas as ações relacionadas a GMP, como atualizações de configuração, exclusões de dados, assinaturas eletrônicas e ajustes de sensores. Essas trilhas precisam de revisões regulares, conforme descrito na orientação de integridade de dados da MHRA e no Anexo 11 da EU GMP.
Para sistemas de produção de carne cultivada, esses controles são particularmente importantes, pois as condições ambientais têm um impacto direto na viabilidade celular.
Uma vez que esses controles estejam em vigor, o sistema deve ser validado e as mudanças gerenciadas cuidadosamente para garantir a conformidade contínua.
Procedimentos de Validação e Controle de Mudanças
Sistemas de monitoramento devem passar por validação através das etapas IQ, OQ e PQ para verificar a precisão dos sensores, funcionalidade dos alarmes, integridade dos dados, processos de backup e trilhas de auditoria. Alternativamente, pode-se usar uma abordagem de ciclo de vida alinhada com os princípios do GAMP 5 e o Anexo 11.
O Anexo 1 das BPF da UE (revisão de 2022) exige que os sistemas de monitoramento ambiental sejam "adequadamente qualificados e validados" e determina que os registros eletrônicos atendam aos padrões do Anexo 11 para integridade, segurança e rastreabilidade. Esses requisitos estabelecem a base para qualquer instalação em conformidade com as BPF.
Qualquer modificação que possa afetar a integridade dos dados, a funcionalidade dos alarmes ou a rastreabilidade deve passar por um processo formal de controle de mudanças. Mesmo atualizações aparentemente menores, como patches de software, podem interromper trilhas de auditoria e não devem ser implementadas sem uma avaliação prévia de impacto.
Diferentes tipos de dados exigem controles GMP personalizados para garantir relatórios precisos e oportunos.
Tipos de Dados e Requisitos de Conformidade Comparados
Cada tipo de dado no monitoramento de salas limpas possui requisitos específicos para manter a conformidade com GMP.A tabela abaixo descreve os controles principais para vários tipos de dados:
| Tipo de dado | Modo de monitoramento | Controles principais de GMP | Base de limite |
|---|---|---|---|
| Contagem de partículas não viáveis | Contínuo ou frequente durante operações em Grau A/B; rotineiro em outros graus | Contadores de partículas validados; captura automática de dados; com alarme; calibração rastreável a padrões; trilha de auditoria para mudanças de configuração | Limites de classe ISO 14644-1; orientação do Anexo 1 para zonas de Grau A/B |
| Pressão diferencial | Contínuo; com alarme | Transmissores de pressão calibrados; gravação automática com registro de data e hora; reconhecimento de alarmes registrado; manter diferencial de 10–15 Pa entre zonas | Anexo 1; design de classificação de sala específico da instalação |
| Temperatura e umidade relativa | Contínuo para processos críticos; periódico em outros locais | Sondas calibradas; captura automática de dados; análise de tendências; limites de alarme baseados nas necessidades do processo e regulatórias | Conhecimento do processo; orientação regulatória; sensibilidade do produto |
| Microrganismos viáveis no ar | Intermitente (amostragem ativa do ar); frequência aumentada para operações críticas | Amostradores qualificados; procedimentos de amostragem controlados; cadeia de custódia para laboratório; resultados vinculados ao lote e localização; registros prontos para investigação | Limites microbianos do Anexo 1 da EU GMP por grau |
| Resultados de contato de superfície | Periódico; pós-limpeza e pós-operação | Métodos de amostragem controlada; rastreabilidade de laboratório; resultados revisados em relação aos limites específicos de grau; vinculados aos registros de limpeza | EU GMP Anexo 1; POPs da instalação |
Cada tipo de dado requer critérios de aceitação definidos, cronogramas de revisão regular, políticas de retenção e um processo de escalonamento para desvios.Aplicar padrões de revisão uniformes a todos os tipos de dados é um erro comum que os reguladores estão examinando cada vez mais. Adaptar o processo de revisão às necessidades específicas de cada tipo de dado garante conformidade e eficiência operacional.
Relatórios, Revisão e Ações Corretivas
Criando Relatórios Conformes
Relatórios alinhados com os padrões ALCOA+ devem ser completos, precisos e acessíveis para auditorias. Um relatório de monitoramento de sala limpa em conformidade com GMP deve ser conciso, verificável e capaz de apoiar decisões de liberação de lote enquanto demonstra controle ambiental. No mínimo, esses relatórios devem:
- Abranger o período e o escopo de monitoramento.
- Resumir as atividades de amostragem em comparação com o cronograma planejado.
- Declarar claramente se algum limite de alerta ou ação foi excedido.
A análise de tendências é um componente chave desses relatórios, empregando ferramentas estatísticas como gráficos de controle, médias móveis e taxas de excursão por 100 amostras para identificar mudanças graduais. Por exemplo, uma tendência mensal mostrando um aumento constante nas contagens viáveis perto de uma linha de colheita de biorreator fornece muito mais insight do que um único evento fora do limite. Adicionar anotações, como atividades de manutenção, ajustes de processo ou mudanças de pessoal, torna os dados mais fáceis de interpretar e mais prontos para auditoria.
As revisões de trilhas de auditoria são outro passo crítico, exigindo que o pessoal treinado documente suas descobertas meticulosamente. Isso inclui registrar quem revisou eventos específicos do sistema, observar quaisquer anomalias e detalhar ações de acompanhamento, tudo dentro de um registro assinado e datado.
A frequência dos relatórios deve estar alinhada com o risco associado. Por exemplo:
- Relatórios associados a lotes são criados para cada execução de produção.
- Os resumos de monitoramento ambiental de rotina são tipicamente semanais ou mensais.
- Relatórios de tendências são preparados mensalmente ou trimestralmente para identificar sinais iniciais de desvio.
A frequência de relatórios escolhida deve ser justificada nos procedimentos operacionais padrão (POPs) e consistentemente seguida. Esses protocolos também fornecem a base para iniciar ações corretivas quando desvios são identificados.
Abordando Falhas de Conformidade
Quando ocorrem desvios, uma resposta estruturada e rastreável é essencial. Cada desvio deve ter um identificador único, uma descrição clara e uma avaliação de risco que avalie tanto o impacto no produto quanto a integridade dos dados. Os desvios também devem ser classificados (menor, maior ou crítico) e vinculados a lotes ou execuções de produção específicos para avaliar se a liberação do lote é afetada ou se testes adicionais são necessários.
O framework CAPA (Ação Corretiva e Preventiva) é central para abordar falhas de GMP. Um CAPA eficaz requer mais do que atribuir eventos a "erro humano". As orientações da EMA e PIC/S enfatizam:
"falha em investigar adequadamente desvios críticos, resultados OOS e questões de integridade de dados" é uma causa recorrente de ações de fiscalização.
Ferramentas de análise de causa raiz, como os 5 Porquês ou diagramas de espinha de peixe, são inestimáveis para descobrir problemas sistêmicos - sejam eles relacionados a lacunas procedimentais, treinamento insuficiente ou fraquezas em controles técnicos. As ações corretivas devem abordar os riscos identificados durante a captura e armazenamento de dados.
Cada CAPA deve incluir critérios de eficácia mensuráveis. Por exemplo: "nenhuma excursão viável repetida de Grau B acima do nível de ação por seis meses." Além disso, revisões de acompanhamento são essenciais para garantir que esses critérios sejam atendidos.Métricas comuns de CAPA incluem:
- O número de ações abertas.
- Tempo médio para encerramento.
- Porcentagem de ações concluídas no prazo.
- Taxa de desvios repetidos, que serve como um forte indicador da eficácia do CAPA.
Uma revisão de cartas de advertência de GMP de 2015 a 2019 revelou que 65–70% das citações de integridade de dados resultaram de investigações inadequadas, documentação ausente ou falhas em revisar e relatar dados corretamente [2]. Isso destaca a importância de relatórios robustos e uma estrutura CAPA responsiva como evidência de uma instalação bem controlada.
Manutenção da Conformidade GMP em Instalações de Carne Cultivada
Para garantir a segurança e a qualidade na produção de carne cultivada, as instalações devem adaptar os controles GMP estabelecidos para atender aos desafios específicos deste campo emergente.Como a integridade dos dados de salas limpas desempenha um papel crítico na manutenção da segurança do produto, refinar as práticas de GMP para carne cultivada é essencial.
Personalizando Práticas de GMP para Carne Cultivada
Estruturas de GMP como o Anexo 1 da UE, originalmente criadas para produtos farmacêuticos, exigem ajustes para abordar os riscos únicos na produção de carne cultivada. Uma avaliação formal de riscos, como uma análise no estilo FMEA ou HACCP, fornece uma base sólida para alinhar os princípios de GMP com cada estágio da produção. Operações críticas como descongelamento de banco de células, inoculação de biorreator, expansão celular e colheita exigem classificações adequadas de salas limpas, protocolos de vestimenta e monitoramento ambiental conforme especificado no Anexo 1.Enquanto isso, tarefas a jusante, como manuseio de andaimes e embalagem, podem aderir aos padrões de higiene de grau alimentício GMP sob Regulamento (CE) nº 852/2004, desde que a rastreabilidade e a integridade dos dados permaneçam intactas durante todo o processo [6] [9][14].
As estratégias de monitoramento ambiental devem se concentrar em organismos relevantes para carne cultivada e segurança alimentar, em vez de se concentrarem apenas em patógenos farmacêuticos tradicionais. A amostragem deve ser priorizada em áreas de alto risco, como aquelas próximas a biorreatores abertos, zonas de preparação de meios e estações de manuseio de andaimes. Esses locais devem ser selecionados com base em padrões documentados de fluxo de ar e análises de movimento de pessoal [9][10].
Dado o alto volume de dados gerados por biorreatores de carne cultivada, os sistemas devem ser capazes de capturar, registrar o tempo e armazenar esses dados com segurança em um repositório validado. O arquivo de dados brutos original do instrumento deve sempre ser identificado como o registro principal para garantir a conformidade [7][8].
Os protocolos de limpeza e desinfecção também exigem consideração cuidadosa. Resíduos considerados aceitáveis em ambientes farmacêuticos podem interferir na adesão ou diferenciação celular na produção de carne cultivada. Os dados de verificação para agentes de limpeza devem ser coletados e mantidos como parte do programa de monitoramento ambiental [3][4].
Usando Recursos da Indústria Como Cellbase
Plataformas de aquisição especializadas são inestimáveis para atender às necessidades específicas de instalações de carne cultivada. Equipamentos de sala limpa compatíveis com GMP devem atender aos padrões de proteção contra entrada e limpeza, além de integrar-se perfeitamente com sistemas de dados validados. Os fornecedores devem fornecer especificações detalhadas, incluindo capacidades de trilha de auditoria, formatos de exportação de dados, configurações de alarme e procedimentos de calibração, juntamente com o equipamento.
Ao adquirir sistemas através de
- Recursos de integridade de dados: trilhas de auditoria seguras, registros com carimbo de data e hora, permissões baseadas em funções e logins de usuário exclusivos
- Compatibilidade do sistema: suporte para protocolos de comunicação padrão e APIs para armazenamento centralizado de dados
- Calibração e manutenção: disponibilidade de documentação abrangente
- Suporte à qualificação: modelos IQ/OQ fornecidos pelo fornecedor para validação simplificada
- Adequação para sala limpa: materiais e designs que facilitam a limpeza
Solicitar a documentação do fornecedor no início do processo de aquisição pode ajudar a evitar possíveis problemas de qualificação no futuro [3][4].
Conclusão
Recapitulação dos Pontos Principais
A conformidade com GMP na gestão de dados de salas limpas gira em torno de demonstrar controle - sobre processos, registros e as decisões informadas por esses dados. Se um registro não for confiável, o processo que ele documenta torna-se igualmente questionável. Este princípio se aplica universalmente, seja lidando com registros de monitoramento ambiental, saídas de biorreatores, relatórios de desvios ou certificados de calibração.
Quatro temas centrais emergiram ao longo desta discussão. Primeiro, integridade de dados, guiada pelos princípios ALCOA+, é a pedra angular da documentação de salas limpas em conformidade. Segundo, gestão do ciclo de vida garante que os dados sejam registrados com precisão, revisados prontamente, armazenados com segurança e retidos pelo período necessário. Terceiro, monitoramento validado e controlado por mudanças formam a base técnica que nenhum SOP pode substituir.Conforme destacado pela análise da MHRA das inspeções de GMP de 2016 a 2021, as deficiências comuns continuam a incluir registros incompletos e revisões insuficientes de trilhas de auditoria [1]. Finalmente, relatórios precisos e rastreáveis garantem que os dados brutos possam ser vinculados a decisões de lote, investigações e ações corretivas, atendendo às expectativas regulatórias.
Para instalações de carne cultivada, esses princípios assumem ainda mais importância. O desafio de combinar fluxos de trabalho no estilo de P &D com controles em nível de produção exige uma governança de dados robusta para unir ambos os ambientes operacionais. A gestão adequada de dados em salas limpas não apenas garante consistência e reprodutibilidade, mas também prepara as instalações para a ampliação, ao mesmo tempo que demonstra a segurança do produto para reguladores, investidores e consumidores.
O conselho mais prático? Aborde os riscos conhecidos antes que os auditores os destaquem.Vulnerabilidades como sistemas híbridos de papel-eletrônico, logins de usuário compartilhados, revisões de dados atrasadas e armazenamento local descontrolado são previsíveis e evitáveis. Resolver proativamente essas questões é muito mais eficaz - e menos custoso - do que reconstruir um rastro de dados após um incidente de qualidade.
Para equipes que buscam equipamentos de monitoramento, sensores ou infraestrutura adaptada a essas necessidades,
Perguntas Frequentes
Como o ALCOA+ pode ser demonstrado nos registros diários de sala limpa?
Para aplicar os princípios ALCOA+ nos registros diários de sala limpa, assegure o seguinte:
- Atribuível: Identifique claramente a pessoa responsável, incluindo carimbos de data e hora para cada entrada.
- Legível: Os registros devem ser fáceis de ler e livres de ambiguidade.
- Contemporâneo: Documentar as informações no momento em que a atividade ocorre.
- Original: Manter a primeira gravação dos dados, não cópias ou transcrições.
- Preciso: Garantir que todas as entradas reflitam os dados verdadeiros sem erros.
- Completo: Incluir todos os dados e metadados relevantes sem omissões.
- Consistente: Manter uma ordem lógica e sequencial nos registros.
- Duradouro: Usar formatos e materiais adequados para preservação a longo prazo.
- Disponível: Manter os registros acessíveis para revisão ou auditorias quando necessário.
Esses passos são críticos para garantir a conformidade com Boas Práticas de Fabricação (GMP) na gestão de dados de salas limpas.
Quais são os principais riscos à integridade dos dados em sistemas híbridos de papel e eletrônico?
Armazenar informações em vários locais introduz complicações na verificação da precisão dos dados. Além disso, a entrada manual de dados aumenta o risco de erros humanos, enquanto sistemas mal controlados ou independentes deixam os registros vulneráveis à manipulação ou exclusão. Esses problemas destacam a necessidade de práticas robustas de gerenciamento de dados para manter a conformidade e preservar a integridade dos dados.
Que evidências os inspetores esperam para a validação do sistema de monitoramento e controle de mudanças?
Os inspetores frequentemente pedem por evidências documentadas que demonstrem a validação do sistema. Isso inclui testar parâmetros críticos como:
- Integridade do filtro HEPA: Garantir que os filtros atendam aos padrões de desempenho exigidos.
- Fluxo de ar e diferenciais de pressão: Verificando se estão dentro de faixas aceitáveis para manter ambientes controlados.
- Dados de monitoramento ambiental: Demonstrando que a instalação atende aos requisitos de limpeza e controle de contaminação.
Além dos testes de validação, manter registros de atividades de controle de mudanças é igualmente importante. Isso abrange ações como substituições de filtros ou modificações na instalação, que ajudam a provar que o sistema continua a funcionar conforme esperado e está em conformidade com os padrões regulatórios.










