

Os registros de lote são críticos para conformidade e segurança do produto. Eles documentam cada etapa da produção, garantindo que os padrões regulatórios sejam atendidos. Para os produtores de carne cultivada, manter a esterilidade e registros detalhados é inegociável. Inspeções da FDA frequentemente destacam problemas como dados ausentes, revisões incompletas e ações corretivas inadequadas, que podem levar a advertências ou interrupções.
Pontos Principais:
- Registros de Lote: Dois tipos - Registro Mestre de Lote (MBR) (a "receita") e Registro de Produção de Lote (BPR) (a "execução").
- Problemas Comuns: Erros humanos (50% dos problemas), verificações em processo ausentes, revisões incompletas e sistemas CAPA (Ação Corretiva e Preventiva) inadequados.
- Padrões da FDA: A adesão aos princípios ALCOA+ (Atribuível, Legível, Contemporâneo, Original, Preciso, Completo, Consistente, Duradouro, Disponível) é obrigatória.
- Soluções: Auditorias independentes, registros eletrônicos de lote e verificação rigorosa de fornecedores podem minimizar erros e melhorar a conformidade.
Empresas de carne cultivada como UPSIDE Foods estabeleceram um padrão ao garantir documentação detalhada, rastreabilidade de insumos e medidas corretivas rápidas. Ao aprender com essas práticas, os produtores podem evitar armadilhas regulatórias e manter altos padrões de qualidade.
Guia Abrangente de Documentação e Manutenção de Registros para Conformidade com a FDA em Ciências da Vida
sbb-itb-ffee270
Problemas Comuns na Documentação de Registros de Lote
Relatórios de inspeção da FDA consistentemente destacam um problema recorrente: desvios na revisão de registros de produção estão entre as principais deficiências de GMP citadas por reguladores [7]. Para os produtores de carne cultivada, essas deficiências vão além de meros erros administrativos - elas colocam em risco a capacidade de demonstrar condições estéreis sustentadas. Esses problemas aparecem de várias formas, como ilustrado nos exemplos abaixo.
Revisões Incompletas e Não Conformidades
Um problema comum é a falha das Unidades de Controle de Qualidade em revisar minuciosamente os registros de lote. Em vez de serem uma parte integral do processo de liberação, as revisões muitas vezes ocorrem de forma reativa - apenas depois que um problema de produto já surgiu [7]. Essa abordagem deixa lacunas significativas nos registros de produção.
Por exemplo, Davis City Pharmacy recebeu uma observação 483 da FDA devido a registros de lote faltando detalhes críticos, como quantidades de componentes, etapas operacionais e iniciais do pessoal. Da mesma forma, CAPS foi citada por falta de assinaturas necessárias e verificações de revisores em entradas chave [3]. Essas falhas não são incidentes isolados; estudos mostram que cerca de 52% das violações de documentação aumentam quando sistemas robustos de gerenciamento de bioprocessos estão ausentes [3].
"Não é a complexidade do processo que desencadeia citações - é a inconsistência, a incompletude e a supervisão inadequada." - Serviços de Consultoria em Auditoria GXP & [5]
Registros de Verificação em Processo Ausentes
Outra deficiência frequente é a ausência de documentação adequada para verificações em processo. Esses registros são cruciais, especialmente em pontos de controle críticos em operações assépticas. Por exemplo, o Centro de Manipulação Estéril Nephron enfrentou citações por não documentar etapas essenciais de vestimenta e procedimentos assépticos em seus registros de lote [3]. Para os produtores de carne cultivada, onde a esterilidade é primordial, tais omissões tornam impossível confirmar a conformidade com medidas de controle de contaminação.
A Amphastar também foi sinalizada por não investigar ou documentar variações inesperadas de rendimento ou discrepâncias de produção [3]. Os riscos de tais negligências são evidentes. Em um caso, uma instalação farmacêutica não identificada em 2024/2025 foi encontrada armazenando registros de lotes concluídos em prateleiras abertas e mesas. Investigadores da FDA descobriram páginas faltantes, incluindo sete de um único registro, e uma seção inteira de "Solução de Síntese" ausente de outro [6].
Falhas de CAPA e Verificação de GMP de Fornecedores
Além dos erros de documentação, a ausência de processos corretivos eficazes e verificação de fornecedores compromete ainda mais a confiabilidade dos registros de lotes.Quando ocorrem desvios de produção sem um relatório de Ação Corretiva e Preventiva (CAPA) correspondente, a integridade dos registros de lote é comprometida [7]. Por exemplo, Eugia Pharma Specialities Limited, inspecionada entre 22 de janeiro e 2 de fevereiro de 2024, recebeu um FDA 483 por não revisar adequadamente as discrepâncias. Seu sistema CAPA ineficaz e investigações incompletas levaram a problemas de produção repetidos, forçando uma reformulação completa de seus procedimentos de investigação e CAPA [9].
Da mesma forma, durante uma inspeção de 26 de setembro a 25 de outubro de 2023, a Stokes Healthcare Inc. demonstrou uma gestão de discrepâncias deficiente. A empresa não conseguiu estender as investigações a todos os lotes afetados e atrasou a conclusão de suas análises [9].
"Uma discrepância sem um relatório CAPA ou de desvio correspondente? Isso é uma falha de conformidade." - GXP Auditing & Serviços de Consultoria [5]
Problemas de verificação de fornecedores adicionam outra camada de complexidade. Empower Clinic Services LLC foi citada durante uma inspeção de 18 de julho a 5 de agosto de 2022 por procedimentos inadequados de controle de qualidade, incluindo qualificações insuficientes de fornecedores e processos de investigação deficientes [9]. Para produtores de carne cultivada, que dependem de meios de crescimento, linhas celulares e outros insumos críticos, garantir a conformidade GMP dos fornecedores é vital para manter a integridade dos registros de lote.
Requisitos da FDA para Registros de Lote
As regras da FDA para registros de lote giram em torno de 21 CFR Parte 117, que estabelece a base para a segurança alimentar.Quando se trata de carne cultivada, onde manter a esterilidade durante a fase de cultura celular é crucial, a documentação muitas vezes precisa atender aos padrões mais rigorosos de Parte 111 ou Parte 211, além da Parte 117 [10][14]. Isso destaca como a documentação precisa é essencial para garantir a segurança e eficácia da produção de carne cultivada.
Padrões Principais para Registros de Lotes
Cada lote requer dois documentos principais:
- Registro Mestre de Lote (MBR): O modelo aprovado que descreve o processo de produção.
- Registro de Produção de Lote (BPR): Um registro detalhado do que realmente acontece durante a execução da produção [12][2].
O BPR deve incluir detalhes como números de lote ou batch, detalhes do equipamento, datas de limpeza, identificadores de componentes, medições exatas e comparações de rendimentos reais versus teóricos [10][14].
"O registro de produção do lote deve seguir com precisão o registro mestre de fabricação apropriado e você deve realizar cada etapa na produção do lote." – 21 CFR 111.255 [12]
Cada etapa crítica deve ser registrada imediatamente, com as iniciais do executor e do verificador anotadas [10][11]. A FDA exige aderência aos princípios ALCOA(+), o que significa que os registros devem ser Atribuíveis, Legíveis, Contemporâneos, Originais e Precisos - assim como Completos, Consistentes, Duradouros e Disponíveis [1].
Se houver qualquer desvio do Registro Mestre de Fabricação, ele deve ser investigado minuciosamente. Isso inclui documentar o problema, realizar uma análise de causa raiz e implementar um Plano de Ação Corretiva e Preventiva (CAPA) [8] [1]. Avaliações iniciais de desvios devem ser registradas dentro de 24–48 horas após a detecção [8]. Para instalações que utilizam sistemas eletrônicos, a conformidade com 21 CFR Parte 11 é obrigatória. Isso inclui assinaturas eletrônicas validadas e trilhas de auditoria seguras e com registro de data e hora [8] [1].
Procedimentos de Retenção e Revisão de Registros
Processos adequados de retenção e revisão de registros são críticos para manter a conformidade e garantir a segurança do produto.Na produção estéril, como a de carne cultivada, cada detalhe nos registros de lote deve passar por uma revisão meticulosa. A equipe de Controle de Qualidade (QC) é responsável por revisar todos os registros de lote, monitorar os resultados e testar os dados antes que um lote possa ser aprovado para distribuição [10] [13].
"Todos os registros de produção e controle de produtos farmacêuticos devem ser revisados e aprovados pela unidade de controle de qualidade antes que um lote seja liberado ou distribuído." – 21 CFR 211.192 [2]
Os fabricantes geralmente visam completar 95% das revisões de lote dentro de 30 dias da produção [2] . No entanto, para os processos estéreis mais complexos envolvidos na carne cultivada, as revisões geralmente levam 7–10 dias, com instalações de alto desempenho alcançando tempos inferiores a 7 dias [2]. Sistemas eletrônicos de registro em lote podem acelerar significativamente essas revisões, como aqueles integrados em sistemas de produção de carne cultivada, - reduzindo o tempo pela metade em comparação com métodos baseados em papel - desde que sejam validados para atender aos requisitos da Parte 11 e manter a integridade dos dados [1].
O que as Empresas de Carne Cultivada Aprovadas pela FDA Fizeram Certo
As empresas de carne cultivada aprovadas pela FDA estabeleceram um padrão elevado ao adotar práticas que abordam desafios de documentação e atendem a rigorosos padrões de segurança.
Quando a UPSIDE Foods se tornou a primeira empresa de carne cultivada a passar pela consulta pré-mercado da FDA em novembro de 2022, eles estabeleceram um modelo para a indústria.A FDA emitiu uma carta de "nenhuma pergunta adicional" após revisar minuciosamente seu processo de produção, que incluiu o estabelecimento de linhagem celular, bancos de células, controles de fabricação e todos os componentes e insumos [16]. Essa conquista destacou a importância da documentação detalhada para atender aos rigorosos requisitos da FDA.
Atendendo aos Padrões de Esterilidade e Conformidade
A conquista notável da UPSIDE Foods foi sua abordagem exaustiva à rastreabilidade de insumos. Cada componente de produção foi cuidadosamente documentado, garantindo uma cadeia clara de responsabilidade desde a linhagem celular inicial até o produto final [16]. Esse nível de transparência permitiu que os revisores da FDA rastreassem cada etapa do processo de fabricação, confirmando que todos os padrões de segurança foram consistentemente atendidos.
"A consulta pré-mercado da FDA com a empresa incluiu uma avaliação do processo de produção da empresa e do material celular cultivado feito pelo processo de produção, incluindo o estabelecimento de linhas celulares primárias e imortalizadas e bancos de células, controles de fabricação, e todos os componentes e insumos." – U.S. Food and Drug Administration [16]
Outras empresas bem-sucedidas seguiram o exemplo implementando documentação detalhada de processos assépticos. Isso incluiu etapas críticas como procedimentos de vestimenta e operações de manuseio estéril [3]. Ao contrário das falhas de documentação anteriores, essas empresas empregaram sistemas de revisão em camadas, envolvendo verificações de operadores, supervisão de produção e revisões de unidades de qualidade, para detectar possíveis erros antes da liberação do lote [15]. Sistemas de registro eletrônico de lote também desempenharam um papel crucial, impondo aprovações obrigatórias em cada etapa e mantendo trilhas de auditoria imutáveis em conformidade com os requisitos da 21 CFR Parte 11 [3][2].
Essas práticas rigorosas naturalmente se estenderam à forma como as empresas lidavam com desvios e falhas.
Processos CAPA para Falhas de Lote
Quando lotes não atendiam às especificações, empresas aprovadas pela FDA tomavam ações rápidas e sistemáticas. Seus processos de Ação Corretiva e Preventiva (CAPA) incluíam análises formais de causa raiz, avaliações de impacto e ações corretivas claramente documentadas [3]. Quaisquer desvios eram geridos dentro de uma estrutura integrada de garantia de qualidade, garantindo que todos os problemas fossem minuciosamente investigados, justificados e documentados antes que a produção continuasse [2].
Olhando para o futuro, a integridade dos dados está definida para ser um foco principal das ações de fiscalização da FDA para 2024–2025 [1].
Como Melhorar Suas Práticas de Registro de Lotes
Fortalecer as práticas de registro de lotes requer documentação precisa para abordar falhas comuns frequentemente identificadas durante inspeções da FDA. Aqui estão algumas estratégias para enfrentar desafios chave.
Conduza Auditorias Independentes de Registro de Lotes
Auditorias regulares de terceiros podem descobrir problemas que revisões internas podem não perceber. Comece focando em sistemas críticos como Sistemas de Gerenciamento de Informações de Laboratório (LIMS), Sistemas de Execução de Manufatura (MES) e Planejamento de Recursos Empresariais (ERP). Priorize documentos com alto impacto regulatório, como registros de testes de liberação, dados de estabilidade e registros de produção de lotes.
Um método eficaz é o teste de recuperação de amostras.Selecione aleatoriamente lotes recentes e reconstrua seu histórico de produção e laboratório. Isso pode ajudar a identificar dados ausentes, assinaturas incompletas ou lacunas de documentação que poderiam levar a citações regulatórias. Verifique as trilhas de auditoria geradas pelo sistema com entradas manuais para identificar alterações ou exclusões não autorizadas.
Revise todos os relatórios de Fora de Especificação (OOS) e Fora de Tendência (OOT) do último ano. Avalie se as análises de causa raiz foram completas e se as Ações Corretivas e Preventivas (CAPAs) foram adequadamente implementadas. Vale a pena notar que problemas de documentação representam 21% das cartas de advertência da FDA, enquanto erros humanos contribuem para 50% dos problemas de registro de lotes na fabricação farmacêutica [2].
"Não é a complexidade do processo que desencadeia citações - é a inconsistência, a incompletude e a supervisão inadequada." – GXP Auditing & Consulting Services [5]
Simule inspeções regulatórias por meio de revisões simuladas periódicas. Esta prática ajuda as equipes a reconhecer inconsistências e potenciais problemas de integridade de dados antes de uma auditoria real. Garanta que todos os registros sigam os princípios ALCOA+: Atribuível, Legível, Contemporâneo, Original, Preciso, Completo, Consistente, Duradouro e Disponível.
Uma vez que a integridade da documentação esteja sólida, concentre-se em verificar a qualidade de todos os insumos de produção.
Teste Todos os Insumos para Contaminação Microbiológica
Testes independentes de esterilidade e potência para todos os insumos são essenciais - não confie apenas nos Certificados de Análise (CoAs) dos fornecedores. Isso é particularmente crucial para produtores de carne cultivada, pois a contaminação pode comprometer lotes inteiros.
Por exemplo, em fevereiro de 2013, Central Admixture Pharmacy Services enfrentou citações da FDA devido ao controle microbiano inadequado durante a liberação de lotes de produtos estéreis. A empresa teve que introduzir procedimentos detalhados de controle microbiano em seus Procedimentos Operacionais Padrão (SOPs) [4].
Pontos de verificação microbiana em processo podem prevenir a dependência excessiva de testes de produto final. Incorpore esses pontos de verificação nos SOPs de liberação de lotes e mantenha documentação contemporânea rigorosa. Registre todos os resultados de testes e etapas de fabricação à medida que acontecem para evitar retrocessos ou entradas atrasadas, o que poderia resultar em citações da FDA.
Mantenha arquivos abrangentes de fornecedores, incluindo CoAs, relatórios de auditoria, acordos de qualidade e um histórico de quaisquer desvios relacionados a materiais recebidos.
O fortalecimento das práticas de registro de lotes envolve ainda alinhar processos com padrões estabelecidos como HACCP e GCCP.
Alinhar Registros com os Padrões HACCP e GCCP
Incorporar os princípios do Análise de Perigos e Pontos Críticos de Controle (HACCP) nos registros de lote garante que as variáveis críticas do processo sejam monitoradas e documentadas ao longo da produção. Isso inclui o estabelecimento de pontos de verificação de testes microbiológicos em processo, em vez de depender apenas de testes na fase final.
Para os produtores de carne cultivada, a adesão aos padrões de Boas Práticas de Cultura Celular (GCCP) é igualmente vital. Os registros de lote devem incluir detalhes das manipulações assépticas, procedimentos de vestimenta e monitoramento ambiental vinculados aos critérios de liberação de lote [3][4]. Esses passos ajudam a manter a conformidade e garantir a segurança do produto.
Dados da indústria mostram que 52% das violações de documentação aumentam quando o software adequado de fabricação de lotes não está em vigor [3][4]. Um exemplo: em fevereiro de 2023, o Nephron Sterile Compounding Centre recebeu uma observação da FDA devido à ausência de procedimentos de controle para verificar variáveis críticas do processo antes da liberação do lote [4]. Isso destaca a necessidade de documentação proativa alinhada com padrões reconhecidos.
A transição para Registros Eletrônicos de Lotes (EBR) pode reduzir significativamente os erros de documentação - em até 50% - através da coleta de dados em tempo real e fluxos de trabalho automatizados [2]. Esses sistemas sinalizam resultados de testes microbiológicos ausentes ou revisões incompletas antes que um lote avance, minimizando o erro humano.
"A FDA espera que os registros sejam ALCOA(+): Atribuíveis, Legíveis, Contemporâneos, Originais, Precisos - além de Completos, Consistentes, Duradouros e Disponíveis." – Atlas Compliance [1]
Cada discrepância ou desvio inexplicado nos registros de lote deve estar vinculado a uma investigação formal e ao sistema CAPA. Limite as permissões de escrita e exclusão para proteger a integridade dos dados eletrônicos de testes microbiológicos. Fabricantes competitivos visam revisar e liberar 95% dos lotes dentro de 30 dias após a conclusão da produção [2].
Essas ações não apenas reduzem o risco de citações, mas também estão alinhadas com os rigorosos padrões de documentação destacados em inspeções recentes da FDA.
Biopharma vs Carne Cultivada: Diferenças nos Registros de Lote
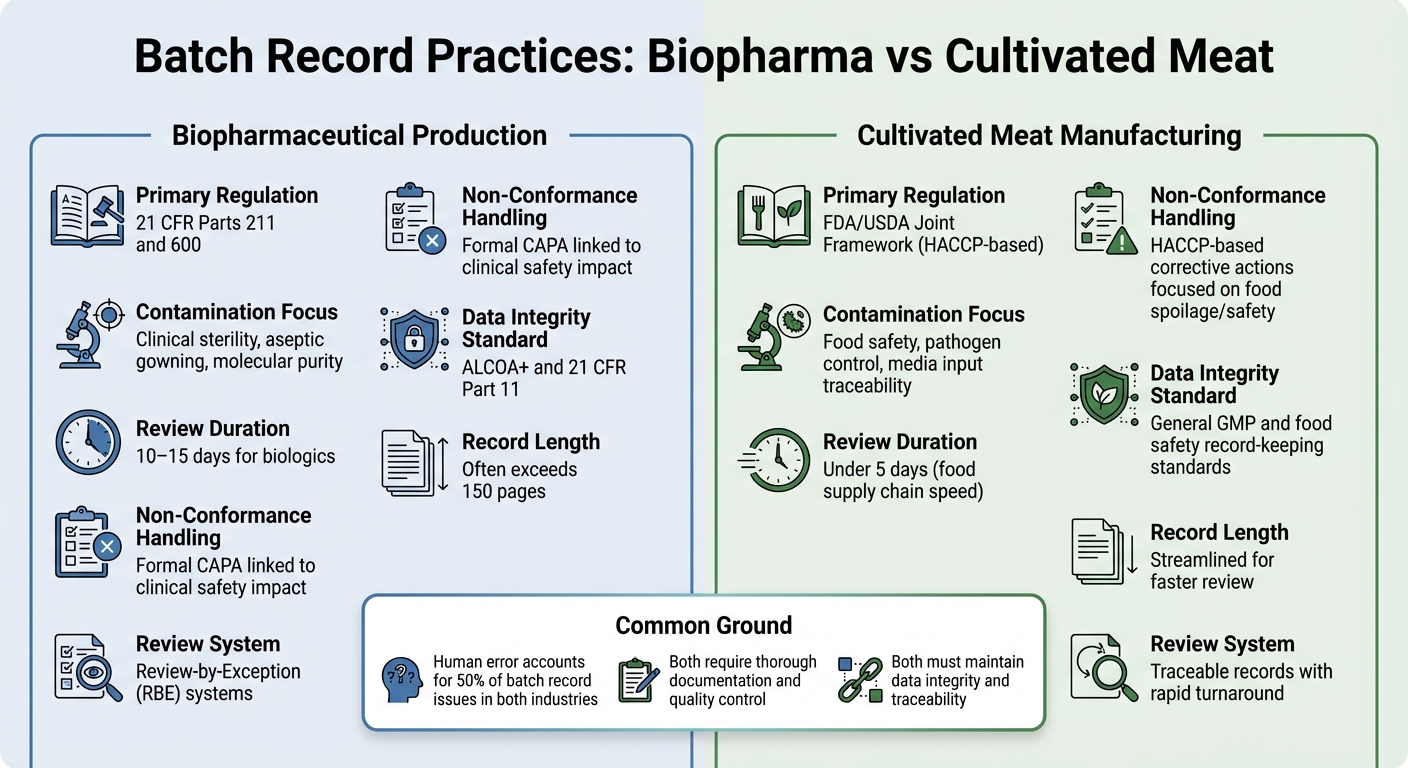
Comparação dos Requisitos de Registros de Lote entre Biofarmacêuticos e Carne Cultivada
Examinar as diferenças nas práticas de registros de lote entre a produção biofarmacêutica e fabricação de carne cultivada oferece uma visão mais clara de como as exigências regulatórias moldam as prioridades de documentação nessas indústrias.
Ambos os setores exigem documentação completa, mas seus frameworks regulatórios e objetivos de controle diferem significativamente. Na biofarmacêutica, os registros de lote são rigidamente regulamentados sob 21 CFR Partes 211 e 600, que exigem que a unidade de Controle de Qualidade revise e aprove todos os registros de produção e controle antes que um lote possa ser liberado [2]. Os produtores de carne cultivada, por outro lado, geralmente seguem os padrões HACCP e GCCP.Esses se concentram mais na segurança alimentar e no controle de patógenos, em vez da esterilidade de grau clínico exigida para biológicos injetáveis.
Os registros de lotes de biofarmacêuticos são frequentemente extensos, às vezes excedendo 150 páginas, e o processo de revisão pode levar de 10 a 15 dias. Para simplificar isso, muitas empresas biofarmacêuticas usam sistemas de Revisão por Exceção (RBE), que resumem desvios chave em um relatório compacto. Enquanto isso, os produtores de carne cultivada visam registros rastreáveis que possam ser revisados em menos de cinco dias, refletindo o ritmo mais rápido da cadeia de suprimento alimentar [2].
O conteúdo desses registros também destaca as prioridades diferentes. As inspeções biofarmacêuticas frequentemente se concentram em detalhes de processamento asséptico, como procedimentos de vestimenta e controles ambientais. Em contraste, os registros de carne cultivada devem enfatizar insumos de mídia e testes microbiológicos para garantir a segurança alimentar.Para carne cultivada, o desafio está em rastrear entradas de mídia complexas, documentar testes microbiológicos para todos os materiais e atender aos limites críticos de segurança alimentar - sem aderir aos requisitos mais rigorosos de esterilidade dos produtos farmacêuticos.
Tendências de Contaminação e Não Conformidade
| Recurso | Produção Biofarmacêutica | Fabricação de Carne Cultivada |
|---|---|---|
| Regulamentação Primária | 21 CFR Partes 211 e 600[2] | Estrutura Conjunta FDA/USDA (baseada em HACCP) |
| Foco em Contaminação | Esterilidade clínica, vestimenta asséptica, pureza molecular[2] | Segurança alimentar, controle de patógenos, rastreabilidade de insumos de mídia |
| Duração da Revisão | 10–15 dias para biológicos[2] | Menos de 5 dias (velocidade da cadeia de suprimento alimentar) |
| Manuseio de Não Conformidade | CAPA formal vinculada ao impacto na segurança clínica [2] | Ações corretivas baseadas em HACCP focadas em deterioração/segurança alimentar |
| Padrão de Integridade de Dados | ALCOA+ e 21 CFR Parte 11 [1] | Padrões gerais de registro de GMP e segurança alimentar |
Embora as taxas de erro humano sejam semelhantes em ambas as indústrias - cerca de 50% dos problemas de registro de lote surgem de erros humanos [2] - as consequências são diferentes.No setor biofarmacêutico, até mesmo uma única desvio não documentado pode ter sérias implicações para a segurança do paciente. Para carne cultivada, os riscos de contaminação estão mais relacionados a patógenos transmitidos por alimentos e deterioração, que podem afetar lotes inteiros de produção.
Conclusão
Os registros de lote servem como o registro oficial para cada lote de produção de carne cultivada - se um passo não for registrado, os reguladores consideram que não foi realizado [6][3]. Isso destaca a importância da documentação precisa e do controle de qualidade rigoroso.
As inspeções da FDA enfatizam que a integridade dos dados deve estar alinhada com os princípios ALCOA+ [1]. As equipes de Controle de Qualidade são obrigadas a revisar e aprovar todos os registros de produção antes que um lote possa ser liberado [2][17], e quaisquer desvios devem ser prontamente investigados com uma análise de causa raiz documentada [2][5]. Embora o erro humano represente 50% dos problemas de registros de lote, revisões em dois níveis e processos estruturados de CAPA (Ação Corretiva e Preventiva) podem ajudar a reduzir esses riscos [2][5].
"Não é a complexidade do processo que desencadeia citações - é a inconsistência, a incompletude e a supervisão inadequada." - GXP Auditing & Serviços de Consultoria [5]
Para superar esses desafios, os produtores de carne cultivada devem focar em auditorias independentes, testes rigorosos de ingredientes seguros para alimentos para contaminação microbiológica e garantir que a documentação esteja em conformidade com os padrões HACCP e GCCP. Implementar sistemas eletrônicos de registro de lotes, validados sob 21 CFR Parte 11 [1], pode minimizar significativamente erros e acelerar os processos de revisão.
O ambiente regulatório exige precisão, mas é navegável.Ao aprender com os erros da biofarmacêutica - como assinaturas ausentes em Qinhuangdao Zizhu Pharmaceutical [17], verificação dupla insuficiente na Terumo Corp [18] , e documentação de desvios inadequada na Torrent Pharmaceuticals [18] - as empresas de carne cultivada podem estabelecer sistemas conformes desde o início. Incorporar essas lições permite conformidade proativa e qualidade consistente. A retenção segura de registros, o relatório oportuno de desvios e a realização de auditorias simuladas realistas garantirão que os registros de lote permaneçam prontos para inspeção e que as execuções de produção sejam totalmente rastreáveis.
Para mais recursos e orientação especializada sobre a manutenção de altos padrões de produção na fabricação de carne cultivada, visite
Perguntas Frequentes
O que um registro de lote deve incluir para carne cultivada?
Um registro de lote para carne cultivada serve como um registro abrangente de todo o processo de fabricação. Ele deve incluir instruções detalhadas de processamento, registros de execução passo a passo, e anotar quaisquer desvios que ocorram durante a produção. Além disso, deve documentar testes em processo e testes de liberação para confirmar que o produto atende aos padrões de segurança, qualidade e regulamentação.
Como podemos comprovar a esterilidade usando registros de lote?
Comprovar a esterilidade através de registros de lote envolve examinar minuciosamente os procedimentos de esterilização documentados, os resultados dos testes e os relatórios de controle de qualidade do meio para garantir que atendam aos requisitos regulamentares.É crucial abordar quaisquer desvios ou testes falhos por meio de investigações detalhadas e CAPAs (Ações Corretivas e Preventivas). Este processo garante que cada etapa tenha sido seguida e quaisquer problemas tenham sido devidamente resolvidos para manter os padrões de esterilidade.
Quando são necessários registros eletrônicos de lote (Parte 11)?
Registros eletrônicos de lote são essenciais sob a Parte 11 quando sistemas eletrônicos são usados para documentar, investigar e justificar desvios de registros de lote. Eles desempenham um papel crítico em garantir a conformidade com 21 CFR Parte 211.192 , protegendo a integridade dos dados, cumprindo os prazos de investigação e garantindo uma supervisão eficaz da gestão.









