

Se você editar primeiro e verificar depois, poderá corrigir uma alteração fora do alvo no clone. Eu manteria o fluxo de trabalho simples: escolha o método de edição de menor risco, mantenha a exposição ao editor curta e, em seguida, teste tanto os locais fora do alvo previstos quanto a estabilidade do clone antes da liberação.
Para engenheiros de bioprocessos, cientistas de cultura celular e equipes de P&D de carne cultivada, o ponto principal é direto. Os sistemas CRISPR ainda podem cortar em locais de correspondência próxima, muitas vezes com 3–6 incompatibilidades toleradas, e esses erros podem ser transferidos para clones de célula única expandidos. O artigo divide o controle de risco em três fases: antes da edição , durante a edição, e após a edição.
Aqui está a lista de verificação completa em termos simples:
-
Escolha a ferramenta de edição de menor risco para o trabalho
- Use edição base ou edição prime quando puderem realizar a edição sem uma quebra de fita dupla
- Use modulação baseada em dCas9 se você só precisar de regulação gênica
- Se precisar de uma nucleasse, comece com uma variante Cas9 de alta fidelidade
-
Proteja o material de partida
- Confirme a identidade da linha celular
- Verifique micoplasma
- Registre o número de passagem
- Sequencie o locus alvo real na linha de trabalho, não apenas o genoma de referência
-
Selecione guias antes do trabalho úmido
- Use ferramentas baseadas em alinhamento e ferramentas baseadas em pontuação para off-target juntas
- Prefira um guia com um perfil off-target mais limpo em vez de um com apenas maior atividade on-target
- Observe o comprimento do guia, 40–60% de conteúdo GC, e sequências homopoliméricas
-
Limite a exposição dentro da célula
- Use RNP ou entrega de mRNA em vez de sistemas plasmidiais ou virais quando possível
- Use a dose mínima eficaz
- Evite estender a persistência do editor apenas para forçar resultados de transfecção
-
Adicione controles extras para casos de maior risco
- Considere nickases emparelhados
- Use sistemas induzíveis, split-Cas9, ou controlados por luz quando o tempo for importante
- Adicione proteínas anti-CRISPR como uma etapa de desligamento quando necessário
-
Valide adequadamente após a edição
- Confirme a edição no alvo primeiro
- Verifique cada local fora do alvo previsto com NGS de amplicon direcionado
- Mova para GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, ou WGS quando o risco do projeto for maior
- Para editores de base ou editores primários, adicione verificações em nível de RNA quando relevante
-
Não libere um único clone apenas com base na sequência
- Compare 2–3 clones independentes
- Use um controle parental não editado
- Remova clones com instabilidade ou desvio de fenótipo
- Libere apenas quando o estado de edição, a triagem fora do alvo e os registros estiverem todos completos
Uma maneira breve de pensar sobre isso: projetar para evitar cortes fora do alvo, entregar para limitar o tempo na célula, depois validar na profundidade que o risco do projeto justifica. Isso é o fio condutor de toda a peça.
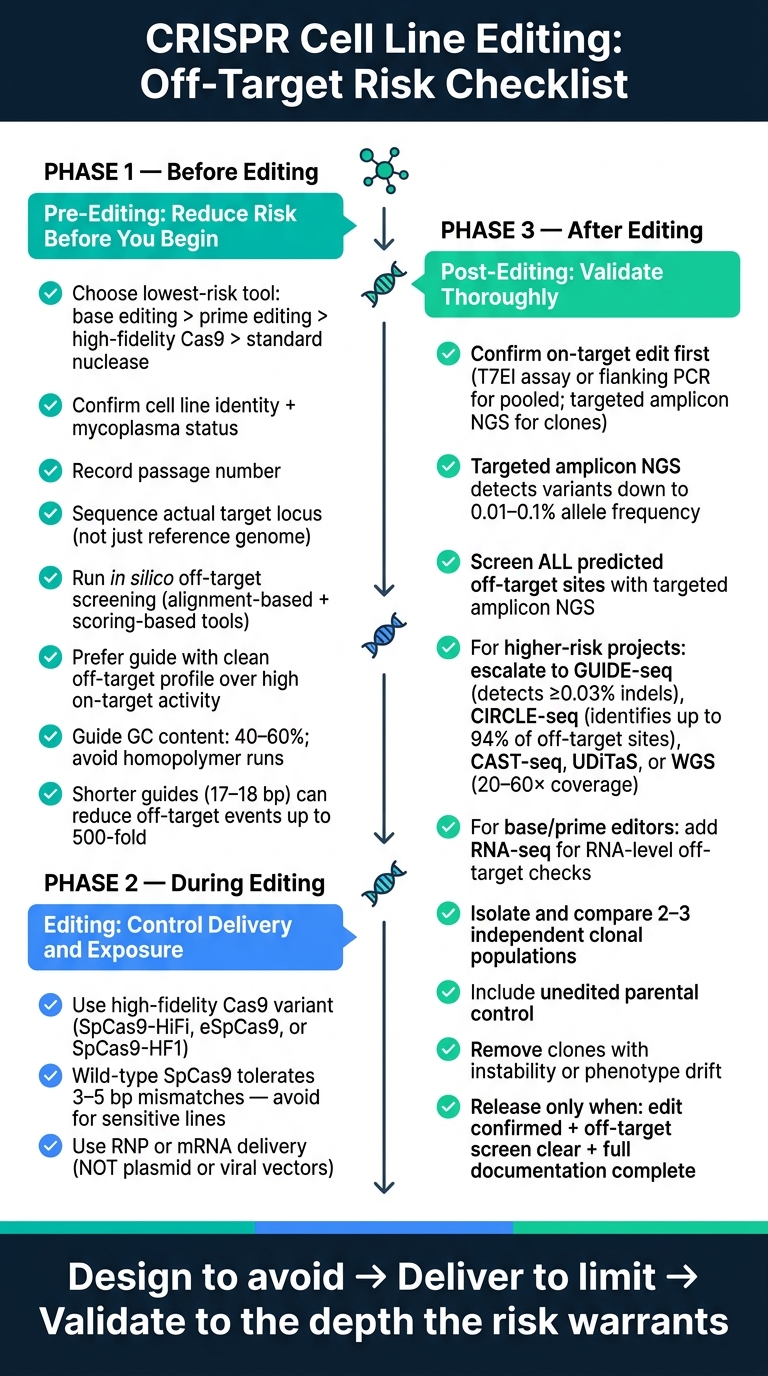
Controle de Risco Fora do Alvo do CRISPR: Lista de Verificação em 3 Fases para Edição de Linhas Celulares
Lista de verificação pré-edição: reduza o risco antes de começar a edição
Defina o objetivo da edição e escolha o método de edição de menor risco
Antes de encomendar um único reagente, tenha clareza sobre o que a edição pretende fazer. Um knockout, um knock-in, uma alteração de nucleotídeo único e modulação transcricional não apresentam o mesmo risco fora do alvo. Eles também não exigem a mesma ferramenta.
A ordem geral de risco é direta. Nucleases formadoras de DSB, como Cas9 e Cas12 , estão no topo do risco porque podem causar grandes deleções, translocações e respostas a danos no DNA [1] [7]. Editores de base e editores primários usam nickases, então evitam DSBs e reduzem o risco de variação estrutural [1][5]. Para modulação transcricional, editores epigenéticos como dCas9 fundido a modificadores transcricionais deixam a sequência de DNA inalterada [1].
A regra prática é simples: use o método menos genotóxico que ainda possa fornecer a edição que você precisa. Para mudanças de nucleotídeo único, CBEs ou ABEs são mais adequados do que HDR, que ainda pode introduzir indels [3] [1]. Para substituições e pequenas inserções ou deleções, a edição primária frequentemente mostra menor atividade fora do alvo do que o CRISPR-Cas9 padrão [1]. Se você precisar usar uma nucleasse, escolha uma variante de alta fidelidade, como SpCas9-HiFi, eSpCas9, ou SpCas9-HF1 [1] [6].
Uma vez que a abordagem de edição esteja definida, fixe a linha celular de trabalho e a sequência alvo exata.
Confirme a identidade da linha celular, histórico e sequência do locus alvo
Se a linha celular estiver mal identificada ou contaminada, o restante do fluxo de trabalho começa a vacilar. Mesmo um RNA guia bem projetado não resgatará material de partida ruim. Verifique a identidade da linha celular antes de iniciar qualquer edição. Ao mesmo tempo, confirme o status de micoplasma e registre o número de passagem atual, já que células de alta passagem podem alterar a estabilidade genômica e a eficiência de edição [1][6].
Igualmente importante, não confie apenas em um genoma de referência.Sequencie o locus alvo exato na linha celular de trabalho. Esse passo ajuda a identificar SNPs ou indels que podem bloquear a ligação do guia ou criar novos locais fora do alvo [1] [6].
Depois disso, passe para o design do guia.
Execute a triagem in silico de fora do alvo antes de selecionar reagentes
Uma vez confirmado o locus alvo, faça a triagem dos RNAs guias candidatos in silico antes de se comprometer com o trabalho de laboratório úmido. Use tanto ferramentas baseadas em alinhamento, como Cas-OFFinder ou FlashFry, e ferramentas baseadas em pontuação, como pontuação CFD ou DeepCRISPR . O primeiro grupo ajuda a encontrar locais genômicos com homologia de sequência. O segundo ajuda a classificar esses locais pela probabilidade de clivagem prevista [1][5].
Ao selecionar guias, um perfil de fora do alvo mais limpo deve superar a eficiência bruta no alvo. Um guia com 70% de eficiência no alvo e sem previsões de fora do alvo é um ponto de partida mais seguro do que um com 90% de eficiência e vários sites de alto risco [6]. Em alguns contextos, encurtar o comprimento do guia de 20 pb para 17-18 pb pode reduzir eventos fora do alvo em até 500 vezes sem muita perda de precisão no alvo [5]. Mire em um conteúdo de GC entre 40% e 60%, e evite sequências de quatro ou mais bases idênticas [6][5].
Dito isso, a triagem in silico tem limites. Ela não considera bem o estado da cromatina, o ciclo celular ou o contexto específico da célula [1][6][4]. Pense nisso como um filtro, não uma prova.Ele restringe o campo, mas não substitui a confirmação experimental.
Leve os sites de maior risco previstos para o plano de edição e validação.
sbb-itb-ffee270
Lista de verificação de edição: escolha do editor de controle, entrega e exposição
Use editores de alta especificidade e RNAs guias bem classificados
Comece com a lista restrita de previsões fora do alvo e use-a para escolher o editor. Na maioria dos casos, uma variante de alta fidelidade do SpCas9 - SpCas9-HiFi, eSpCas9 ou SpCas9-HF1 - é uma escolha melhor do que o SpCas9 do tipo selvagem [6][1] . O SpCas9 do tipo selvagem pode tolerar até três a cinco incompatibilidades de pares de bases, especialmente na região distal ao PAM, e isso cria um risco significativo fora do alvo em linhas celulares sensíveis [3].
Uma regra simples ajuda aqui: use o editor de alta fidelidade menos ativo que ainda entregue a edição pretendida.
Para editores base, rastreie edições de espectador e efeitos fora do alvo de RNA separadamente do risco fora do alvo de DNA [1] [8] . Esses são modos de falha diferentes e precisam de verificações separadas. Se você puder fazer a edição sem quebras de fita dupla, edição base ou edição prime podem se encaixar melhor em fluxos de trabalho de maior risco [1][8].
Uma vez escolhido o editor, o próximo trabalho é manter seu tempo dentro da célula o mais curto possível.
Limite a persistência do editor com entrega transitória e dose mínima eficaz
A persistência do editor é tão importante quanto a escolha do editor.Quanto mais tempo o editor permanece ativo na célula, mais tempo ele tem para agir em locais de baixa probabilidade. Isso faz do formato de entrega um ponto de controle importante.
Use entrega transitória como RNPs ou mRNA, e evite DNA plasmidial ou vetores virais que prolongam a expressão do editor [1] [5] . Na prática, a entrega de RNP deve ser o padrão [6].
A dosagem também é importante. Alta concentração de nucleases aumenta a chance de clivagem em locais fora do alvo de baixa sensibilidade [5]. Use a dose mínima eficaz. Se a eficiência de transfecção for baixa, não adicione mais reagente e espere pelo melhor. Isso geralmente desloca o problema em vez de resolvê-lo.
Adicione salvaguardas de precisão para fluxos de trabalho de maior risco
Alguns fluxos de trabalho precisam de proteções extras. Isso é especialmente verdadeiro para alvos próximos a oncogenes, supressores de tumor, ou em linhas celulares sensíveis ao p53, onde um evento fora do alvo pode ter um custo desproporcional [1][6][3].
Salvaguardas úteis incluem:
- Nickases emparelhadas, que requerem dois cortes próximos. Um único corte fora do alvo geralmente é reparado sem mutação, então o risco fora do alvo cai bastante em comparação com uma configuração padrão de nucleases [4][1].
- Sistemas Cas9 induzíveis, controlados por luz ou divididos, que ajudam a manter a atividade do editor dentro de uma janela restrita quando a entrega é eficiente e a exposição deve ser breve [1].
- Proteínas Anti-CRISPR (Acr), que atuam como um interruptor de desligamento. Essas proteínas Acr, que ocorrem naturalmente, podem desativar o complexo CRISPR-Cas após um intervalo definido, proporcionando um freio molecular na atividade do editor [1].
Lista de verificação pós-edição: detectar eventos fora do alvo e validar clones
Examinar os locais fora do alvo previstos com sequenciamento direcionado
Uma vez concluída a edição, confirme primeiro a alteração pretendida no locus alvo. Para uma primeira análise rápida em células agrupadas, você pode usar um ensaio de clivagem por incompatibilidade, como T7 Endonuclease I, uma digestão de restrição ou um PCR flanqueador.Apenas tenha cuidado com a interpretação: cada um desses métodos tem limites de sensibilidade, especialmente para edições raras ou variantes homozigóticas [9].
Para validação em nível de clone, NGS de amplicon direcionado é o padrão. Ele fornece uma visão quantitativa da frequência alélica e pode detectar variantes até 0,01% a 0,1% [3].
Sequencie cada site off-target previsto com NGS de amplicon direcionado. Esse deve ser o passo padrão de validação.
Escale para ensaios de genoma completo ou estruturais quando o risco do projeto for maior
A triagem site a site nem sempre é suficiente. Se o editor, o locus alvo ou a linha celular sugerirem risco oculto, passe para ensaios que possam detectar eventos que você não previu antecipadamente.
Ensaios de descoberta em todo o genoma como GUIDE-seq e CIRCLE-seq não precisam de listas prévias de sites off-target.O GUIDE-seq pode detectar locais fora do alvo com frequências de indel tão baixas quanto 0,03% [2]. O CIRCLE-seq pode identificar até 94% dos locais fora do alvo in vitro [3]. Esses métodos são úteis quando o contexto do tipo celular pode mascarar a atividade fora do alvo.
Se você está preocupado com grandes rearranjos, leituras padrão de amplicons podem perder o principal problema. Deleções, inversões e translocações precisam de ensaios construídos para mudanças estruturais, como CAST-seq e UDiTaS [1].
O sequenciamento do genoma completo (WGS) é a opção mais ampla. Ele pode detectar indels, variação estrutural e alterações no número de cópias em todo o genoma [1]. A troca é entre profundidade e custo: geralmente precisa de 20–60× cobertura, o que o torna inadequado para triagem de rotina de populações em massa [1].
Use NGS de amplicon direcionado para locais previstos. Mude para ensaios genômicos ou estruturais para projetos de maior risco. Para editores de base ou prime, adicione RNA-seq para verificar efeitos fora do alvo em nível de RNA.
Selecione múltiplos clones independentes e documente os critérios de liberação
Após as verificações de sequência, teste o fenótipo em mais de um clone.
Não avance com um único clone editado. Isole e expanda pelo menos duas a três populações clonais independentes e compare-as com um controle parental não editado [4][9] . Remova clones que mostrem instabilidade ou desvio de fenótipo [3]. Em seguida, confirme a edição pretendida no estado do alelo necessário, seja heterozigoto ou homozigoto, usando NGS de amplicon direcionado [9].
A documentação não é trabalho administrativo no final. É parte do lançamento do clone. Registre o fundo da linha parental, o design do sgRNA, a variante da nucleasse, o método de entrega e todos os resultados de QC [3]. Um clone só deve avançar quando a edição pretendida for confirmada, os locais previstos fora do alvo estiverem claros e o registro completo estiver em ordem.
Edição de Genoma com CRISPR: Como Minimizar Eficazmente os Efeitos Fora do Alvo
Conclusão: um checklist de três fases para edições mais limpas em linhas celulares
Em conjunto, o checklist trata o controle fora do alvo como um processo em etapas, não como uma verificação de QC única. O objetivo é simples: reduzir o risco cedo, limitar a atividade do editor e, em seguida, verificar o resultado.
A profundidade da validação deve corresponder ao risco. Liberar apenas múltiplos clones independentes confirmados no estado de alelo pretendido.
Perguntas Frequentes
Por que não confiar em um clone?
Confiar em um único clone é arriscado. A edição CRISPR não é perfeitamente específica, portanto, pode introduzir mutações fora do alvo não intencionais.
É por isso que as equipes geralmente expandem múltiplas populações clonais. Fazer isso facilita encontrar uma linha que carregue a edição pretendida no alvo sem alterações prejudiciais fora do alvo.
Há outra razão também: as linhas celulares podem mostrar heterogeneidade genética. Sequenciar múltiplos clones ajuda a confirmar que o knockout homozigoto pretendido ou outra modificação no local-alvo está presente em todos os loci-alvo.
Quando o NGS de amplicon é suficiente?
O sequenciamento de próxima geração baseado em amplicon é frequentemente suficiente quando você precisa de uma maneira direcionada e econômica para confirmar potenciais locais fora do alvo sinalizados por ferramentas computacionais ou outros métodos de triagem.
O sequenciamento do genoma completo ainda é a única maneira de quantificar totalmente os efeitos fora do alvo. Mas para muitas aplicações, esse nível de análise simplesmente não é necessário.
Como escolher o editor mais seguro?
Escolha a variante de nucleases CRISPR menos ativa que ainda corte bem o seu local alvo.
Você não pode escolher a melhor variante apenas pela previsão. A única maneira confiável é realizar uma pequena triagem entre variantes de nucleases selecionadas e ler a edição com sequenciamento de próxima geração.
Para carne cultivada R& D, isso lhe dá um caminho prático: comece com uma lista curta de variantes, depois teste as mais fracas passo a passo até encontrar a opção menos ativa que ainda edita o local alvo de forma eficiente.









