

Validering af vækstmedier er et obligatorisk skridt for virksomheder inden for dyrket kød, der søger godkendelse på det britiske marked. Denne proces sikrer produkternes sikkerhed, kvalitet og overholdelse af strenge reguleringsrammer som de britiske Novel Food Regulations (EU 2015/2283). Her er, hvad du behøver at vide:
- Nøglekrav: Vækstmedier skal opfylde standarder for toksikologi, kontaminationskontrol, ernæringskvalitet og allergenicitet.
- Britiske Regler: Food Standards Agency (FSA) kræver overholdelse af HACCP-principper og klassificering under Produkter af Animalsk Oprindelse (POAO).
- Globale Standarder: Mens Storbritannien og EU deler lignende rammer, følger USA CGMP-regler under FD&C Act.
- Valideringsproces: Omfatter grundig testning af sammensætning, renhed, funktionalitet og leverandøroverensstemmelse, sammen med robust dokumentation.
- Støtteinitiativer: Det britiske £1,6 millioner regulatoriske sandkasse, lanceret i 2025, hjælper virksomheder med at opfylde disse standarder.
Korrekt validering sikrer sikkerhed, opbygger tillid og er i overensstemmelse med lovkrav. Artiklen dykker dybere ned i trin-for-trin processen, herunder testmetoder, leverandørkvalifikationer og tips til regulatorisk indsendelse.
Regulatoriske standarder for vækstmedier
Standarder og retningslinjer
Vækstmedier, en kritisk komponent i produktionen af dyrket kød, skal opfylde strenge internationale regulatoriske standarder. Disse standarder varierer på tværs af regioner, hver med specifikke krav til sammensætning, sikkerhed og renhed.
I Storbritannien er vækstmedier reguleret under Novel Food Regulations (assimileret forordning (EU) 2015/2283). Før de godkendes til markedet, kræves en grundig sikkerhedsvurdering [1]. Food Standards Agency (FSA) klassificerer cellekultiverede produkter som produkter af animalsk oprindelse (POAO) under forordning (EF) 853/2004. Denne klassifikation kræver, at producenter implementerer fødevaresikkerhedsstyringssystemer baseret på principperne for Hazard Analysis and Critical Control Points (HACCP) [3]. FSA er også i gang med at udvikle detaljeret teknisk vejledning om sammensætningen af vækstmedier, med yderligere opdateringer forventet [1]. Disse rammer danner grundlaget for mere specifikke reguleringskrav.
I USA er tilgangen anderledes.Komponenter til vækstmedier skal opfylde kravene i den nuværende god fremstillingspraksis (CGMP), som er beskrevet i afsnit 501(a)(4)(B) i Federal Food, Drug, and Cosmetic Act (FD&C Act) [4]. FDA kategoriserer mediekomponenter som "forsyninger og reagenser", der er reguleret af 21 CFR dele 210 og 211. Disse komponenter skal gennemgå kvalitetsverifikation for at forhindre kontaminering [4]. Interessant nok klassificeres syntetiske komponenter af dyrket kødmedie - såsom aminosyrer, vitaminer og salte - ofte som Klasse I medicinsk udstyr under 21 CFR 864.2220, hvilket fritager dem fra krav om forudgående markedsmeddelelse [6][7].
I Den Europæiske Union er det regulatoriske rammeværk tæt på det britiske, da det også følger forordning (EU) 2015/2283.Den Europæiske Fødevaresikkerhedsautoritet (EFSA) overvåger godkendelsesprocessen [1]. Ifølge ICH Q6B-retningslinjerne behandles vækstmediekomponenter, herunder antibiotika, induktorer og andre bestanddele, som procesrelaterede urenheder. Disse urenheder skal kontrolleres og reduceres til acceptable niveauer [5]. Hvor det er muligt, bør hjælpestoffer og reagenser overholde farmakopéstandarder [5].
| Jurisdiktion | Primær Regulering | Klassifikation | Sikkerhedssystem | Medietilsyn |
|---|---|---|---|---|
| Storbritannien (GB) | Assimileret Regulering (EU) 2015/2283 [1] | Produkt af animalsk oprindelse (POAO) [3] | HACCP (Reg 852/2004) [3] | FSA/FSS Sandbox Vejledning [1] |
| Den Europæiske Union / NI | Regulering (EU) 2015/2283 [1] | Produkt af animalsk oprindelse (POAO) [3] | HACCP (Reg 852/2004) [3] | EFSA-godkendelsesproces [1] |
| United States | FD&C Act Section 501(a)(4)(B) [4] | Ny dyrelægemiddel / Fødevarer [4] | CGMP (21 CFR 210/211) [4] | FDA CVM / USDA-FSIS [4] |
Regulatoriske krav til dyrket kød
Producenter af dyrket kød skal sikre, at hver batch af vækstmedier overholder strenge sikkerheds- og kvalitetsstandarder.Vækstmedievalidering er en nøgleaspekt af den bredere reguleringsramme for disse produkter. Under HACCP-principperne (Forordning (EF) 852/2004) identificeres vækstmedier som en primær input og en potentiel kilde til forurening - kemisk, mikrobiologisk eller på anden måde [3]. FSA fremhæver denne bekymring:
"De vigtigste farer i produktionen af cellekultiverede produkter vedrører cellelinjens identitet (og konsistens), farer introduceret under produktionsprocessen (mikrobiologisk forurening, vækstmedier og resterende komponenter i det endelige produkt) og allergener." [3]
Hvis der er ændringer i vækstmedieformuleringen, kræves en øjeblikkelig HACCP-gennemgang [3].I Storbritannien skal validering finde sted før implementering for at sikre nøjagtigheden af flowdiagrammer og effektiviteten af kontrolforanstaltninger [3].
I USA kræver FDA, at alle reagenser og mediekomponenter opfylder strenge kvalitetsstandarder for at undgå introduktion af skadelige stoffer [4]. Leverandører og kontraktlaboratorier skal overholde CGMP-reglerne, og enhver leverandør, der ikke overholder, bør fjernes for at forhindre, at produkter klassificeres som "forfalskede" [4]. FDA understreger vigtigheden af dette:
"Alle nye dyrelægemidler, inklusive ACTP'er, skal fremstilles i overensstemmelse med CGMP for at sikre, at sådanne lægemidler opfylder kravene i Federal Food, Drug, and Cosmetic Act (FD&C Act) med hensyn til sikkerhed." [4]
I øjeblikket deltager flere virksomheder i Storbritanniens regulatoriske sandkasse - såsom BlueNalu, Gourmey, Hoxton Farms, Mosa Meat, Roslin Technologies, Vital Meat, og Vow - samarbejder med FSA for at forfine disse tekniske standarder [1]. Under britiske regler kan virksomheder anmode om op til fem års databeskyttelse for fortrolige oplysninger indsendt under godkendelsesprocessen [1].
Trin til validering af vækstmedier
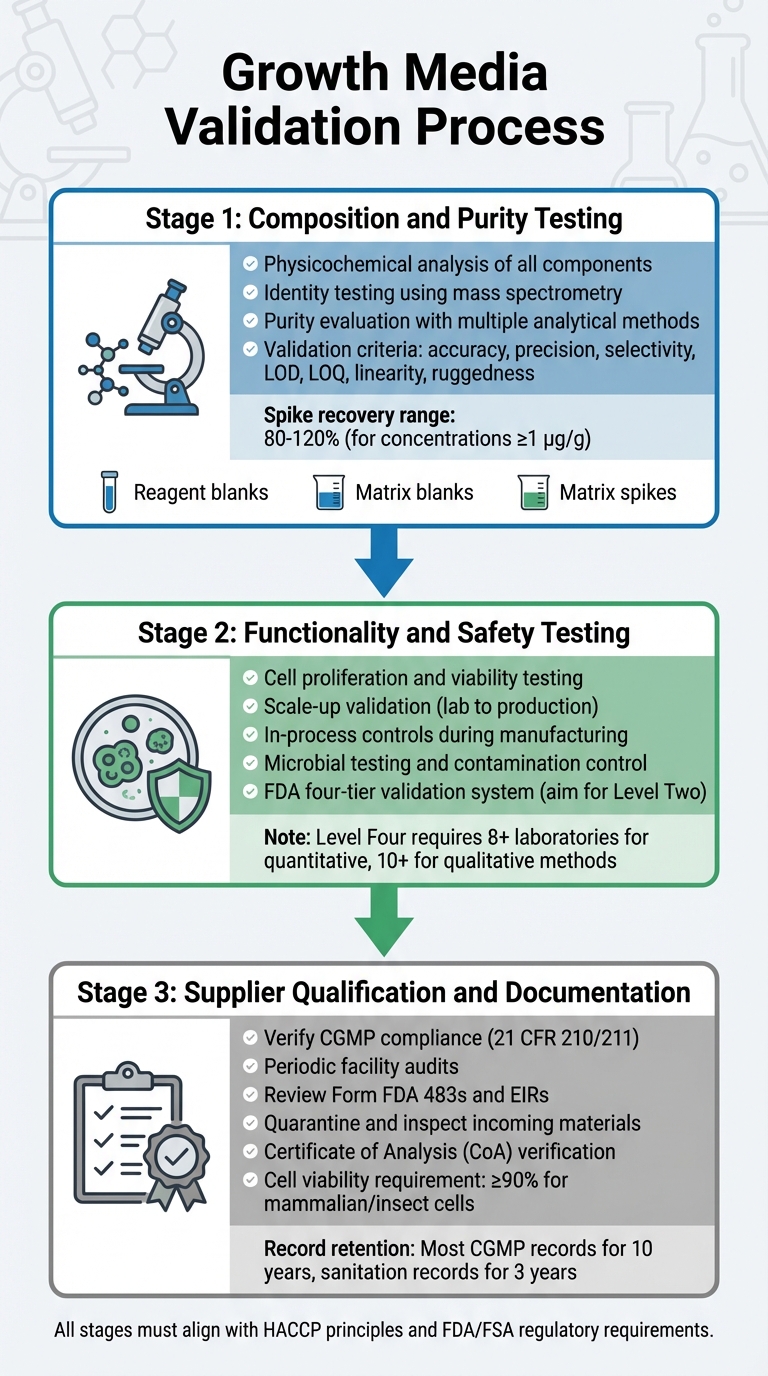
Valideringsproces for vækstmedier til regulatorisk godkendelse af dyrket kød
Validering af vækstmedier involverer en detaljeret proces, der undersøger dets sammensætning, funktionalitet, sikkerhed og leverandørens overholdelse. Hvert trin bygger på det forrige, hvilket sikrer en robust valideringsproces, der er i overensstemmelse med regulatoriske krav. Dette inkluderer testning af sammensætning, funktionalitet og leverandørens overholdelse.
Sammensætning og renhedstest
Den første fase fokuserer på en grundig fysikokemisk analyse af hver komponent. Dette involverer identifikation af den præcise sammensætning, fysiske egenskaber og molekylære struktur af ingredienser som aminosyrer, vitaminer og uorganiske salte [5].For at bekræfte molekylære strukturer anvender identitetstestning meget specifikke metoder, herunder fysikokemiske, biologiske og immunokemiske teknikker. Værktøjer som massespektrometri bruges til at verificere molekylære identiteter gennem deres fragmenteringsmønstre [8].
Renhedsevaluering kræver flere analytiske metoder til at adskille ønskede komponenter fra urenheder. Disse tests skal adressere både procesrelaterede og produktrelaterede urenheder [5]. Analytiske teknikker bør opfylde strenge valideringskriterier, herunder nøjagtighed, præcision, selektivitet, detektionsgrænser (LOD), kvantificeringsgrænser (LOQ), linearitet og robusthed [8]. Valideringsprotokoller bør også inkorporere:
- Reagensblanke for at sikre, at reagenser er fri for analytter.
- Matrix blanks for at bekræfte, at prøveomgivelserne ikke forstyrrer.
- Matrix spikes for at estimere genfinding og nøjagtighed.
For kvantitative metoder ved koncentrationer på 1 µg/g (ppm), er acceptable spike-genfindinger typisk mellem 80% og 120% [8].
For at opretholde konsistens bør producenter etablere interne primære referencematerialer afledt af produktionsrepræsentative partier. Disse tjener som sporbare standarder til kalibrering af arbejdende referencematerialer [5]. Når renhedstestningen er afsluttet, skal mediet demonstrere sin evne til at understøtte effektiv cellevækst og opfylde sikkerhedsstandarder.
Funktionalitet og Sikkerhedstest
Efter at have bekræftet sammensætningen, skal mediet bevise sin effektivitet i at understøtte produktionen af dyrket kød. Dette inkluderer at demonstrere, at celler kan proliferere, opretholde levedygtighed og skalere fra laboratorieforhold til produktionsvolumener. FDA kræver in-process kontrol under fremstillingen, startende fra tidlige stadier som cellepassage og høst, for at sikre produktkonsistens og sikkerhed [4].
Sikkerhedsvalidering involverer streng mikrobiel testning og kontaminationskontrol, som beskrevet i FDA's præ-markedsevalueringer [9].
FDA bruger et fire-niveau system til kemisk metodevalidering, der spænder fra Niveau Et (nødsituation eller begrænset brug) til Niveau Fire (fulde samarbejdsstudier, der opfylder AOAC/ISO standarder) [8].Til rutinemæssig reguleringsprøvning, sigt efter Level Two single-laboratory validation, som inkluderer en omfattende præstationsvurdering [8]. Fuldstændige samarbejdsstudier for kvantitative metoder kræver deltagelse fra mindst otte laboratorier, mens kvalitative metoder har brug for ti [8]. Når mediets ydeevne er valideret, er det vigtigt at sikre, at alle råmaterialer kommer fra overensstemmende leverandører.
Leverandørkvalifikation og dokumentation
Producenter skal arbejde med verificerede, CGMP-overensstemmende leverandører. Leverandører bør opfylde de standarder, der er beskrevet i 21 CFR 210/211 [4]. Verifikation involverer periodiske audits af leverandørfaciliteter for at vurdere overholdelse af kvalitetsprogrammer, procedurer og generel CGMP-overensstemmelse [4].
Før indgåelse af kontrakter, gennemgå en leverandørs overholdelseshistorik, inklusive Form FDA 483s og Establishment Inspection Reports (EIRs) [4]. FDA understreger denne forpligtelse:
"Før indgåelse af en kontrakt, aftale eller anden ordning med en anden virksomhed for at udføre et produktionsskridt for dig, bør du verificere, at virksomheden overholder gældende reguleringsmæssige CGMP." [4]
Alle indkommende materialer skal i karantæne og inspiceres før frigivelse, for at sikre at de opfylder hovedspecifikationerne [10]. Leverandører er forpligtet til at levere et Certificate of Analysis (CoA) eller sporbare, CGMP/GLP-kompatible testresultater [10].For stabile cellelinjer skal dokumentationen inkludere en sporbar kloningshistorik [10]. Pattedyr- eller insektceller kræver typisk mindst 90% levedygtighed for accept i CGMP-projekter [10]. Optegnelser skal opbevares i henhold til regulatoriske retningslinjer [4].
Kontrakter skal tydeligt skitsere CGMP-ansvar og kræve, at leverandører underretter producenter om eventuelle foreslåede ændringer i testkits eller metoder [4]. Hvis testning er outsourcet, skal det sikres, at kontraktlaboratorier bruger validerede analytiske metoder og er FDA-registreret [4].
Forberedelse af dokumenter til reguleringsindsendelse
Når dit vækstmedie er blevet valideret, er det næste skridt at udarbejde et dossier, der demonstrerer overholdelse af alle sikkerheds- og kvalitetsstandarder krævet af FDA og USDA-FSIS. Dette dossier fungerer som et kritisk bindeled mellem validering og reguleringsmæssig overholdelse, hvilket giver myndighederne et klart billede af dit medies sikkerhed og produktionsprocesser.
Nødvendige elementer i et indsendelsesdossier
Dit dossier bør indeholde en detaljeret opdeling af mediekompositionen, der opregner alle aminosyrer, vitaminer, uorganiske salte og vækstfaktorer. FDA-retningslinjer understreger, at gennemgangsprocessen ikke kun evaluerer selve mediet, men hele produktionsarbejdsgangen. Dette inkluderer etablering af cellelinjer og banker, implementering af produktionskontroller og verifikation af alle komponenter og input [11].
Derudover skal dossieret indeholde en grundig sikkerheds- og toksikologisk vurdering, der beviser fødevaresikkerheden af det dyrkede materiale og alle dets input. Inkluder fremstillingskontroloptegnelser, procesvalideringsdata og dokumentation for kvalitetsprogrammer for at demonstrere, at din produktion er konsistent og fri for forureninger.
Du bør også give forsynings- og reagensverifikationsoptegnelser, der viser validering for alle materialer, der anvendes i mediet, inklusive dem, der er fremstillet internt. For produkter reguleret af USDA-FSIS, inkluder HACCP-planer og sanitetsprotokoller. FDA anbefaler at beholde de fleste CGMP-optegnelser i mindst 10 år, mens optegnelser for rengøring og sanitet af faciliteter bør opbevares i mindst 3 år [4]. Dette er i overensstemmelse med leverandørkvalifikationsindsatser, der sikrer, at alle input opfylder CGMP- og lovgivningsmæssige krav.
Dokumentation af facilitetsoverholdelse
Før produktion, forarbejdning eller opbevaring af dyrket kød til menneskeføde, skal faciliteter registreres hos FDA [12]. Din dokumentation skal inkludere en omfattende fødevaresikkerhedsplan, der adresserer fareanalyse (biologisk, kemisk og fysisk), forebyggende kontrolforanstaltninger (såsom sanitet, allergenhåndtering og forsyningskædeforanstaltninger) og tilsynsprocedurer [12].
Mediefyldningssimuleringer er også et nøglekrav. Disse involverer 14-dages inkubation og vækstfremmende test for at bekræfte aseptiske praksisser.Som FDA forklarer:
"Mediefyldningen bør evaluere den aseptiske samling og drift af det kritiske (sterile) udstyr, kvalificere operatørerne og vurdere deres teknik, samt demonstrere at miljøkontrollerne er tilstrækkelige" [2].
Sørg for, at dine optegnelser inkluderer leverandørkvalifikationsdata, såsom tests udført på de første tre partier af medium fra en leverandør for at bekræfte, at de matcher analysecertifikatet. Andre vigtige optegnelser inkluderer miljøkontrollogbøger, udstyrskalibreringsplaner og temperaturovervågningsdata. For USDA-regulerede processer, forbered HACCP-planer, skriftlige sanitære standard driftsprocedurer (SSOP'er) og tilbagekaldelsesprocedurer [12][13].
sbb-itb-ffee270
Brug af Cellbase til indkøb af vækstmedier i overensstemmelse med reglerne
Verificerede leverandører til dyrket kød
Når du har valideret din vækstmedieformulering, er det næste skridt at skaffe komponenter, der opfylder de lovgivningsmæssige standarder. Dette er ikke så simpelt som at bestille fra generiske leverandører. For celle-dyrkede produkter gælder der strenge hygiejneregler, og hver komponent i vækstmediet skal leveres med specifik dokumentation for lovgivningsmæssig godkendelse [3]. Det er her
Indkøbsfunktioner
Platformen tilbyder også gennemsigtig prissætning og en direkte beskedfunktion, der gør det muligt for teams hurtigt at anmode om tilbud, analysecertifikater og andre lovgivningsdokumenter.Ved at konsolidere disse kritiske indkøbsfunktioner i ét system skræddersyet til produktion af dyrket kød, forenkler
Konklusion
Validering af vækstmedier til regulatorisk godkendelse er ikke bare en formalitet - det er et lovkrav for at introducere dyrkede kødprodukter på det britiske marked. Dette indebærer grundig testning for sammensætning og renhed, implementering af en stærk HACCP-plan og opretholdelse af detaljeret dokumentation gennem hele processen.
"Fødevarer må ikke markedsføres, hvis de er usikre. Dette betyder, at de hverken må være skadelige for sundheden eller uegnede til menneskeføde." - Food Standards Agency [3]
The UK Food Standards Agency's £1.6 millioner Regulatory Sandbox fremhæver sit engagement i at arbejde med brancheaktører for at etablere klare tekniske retningslinjer for vækstmediesammensætning [1]. Virksomheder, der prioriterer korrekt validering nu, vil være i en stærkere position, når disse retningslinjer er fuldt defineret.
At opfylde overensstemmelsesstandarder handler ikke kun om at krydse af i regulatoriske bokse - det handler om at opnå forbrugertillid og sikre produktsikkerhed. Streng kvalitetstestning er kernen i både regulatorisk godkendelse og opnåelse af markedsaccept. For at strømline godkendelsesprocessen skal der fokuseres på at opbygge stærke valideringsprotokoller, opretholde nøjagtige optegnelser og samarbejde med pålidelige leverandører. Disse skridt vil ikke kun forenkle godkendelsen, men også bane vejen for større forbrugertillid.
Ofte stillede spørgsmål
Hvad er de vigtigste trin for at validere vækstmedier til regulatorisk godkendelse?
Validering af vækstmedier til regulatorisk godkendelse handler om at bevise, at formuleringen er sikker, pålidelig og egnet til at producere dyrket kød. Her er, hvordan processen normalt ser ud:
- Risikovurdering: Start med at definere den cellelinje, du vil bruge, produktets mål og dets kritiske kvalitetsattributter (som pH eller næringssammensætning). Identificer eventuelle potentielle farer, såsom mikrobiel forurening, og læg planer for at kontrollere disse risici.
- Testning og specifikationer: Sæt klare acceptkriterier for faktorer som sterilitet, renhed og styrke. Brug etablerede testmetoder for at sikre, at disse standarder konsekvent opfyldes.
- Valideringsstudier: Udfør grundig procesvalidering, herunder kvalificering af udstyr og testning af flere batches, for at bekræfte, at resultaterne er reproducerbare og konsistente.
- Stabilitetstest: Kontroller, hvordan mediet holder sig over tid ved at vurdere dets kvalitet gennem hele den tilsigtede holdbarhed under korrekte opbevaringsforhold (typisk 2–8 °C).
- Dokumentation: Saml alt i et omfattende valideringsdossier. Dette bør inkludere alle testresultater og analyser for at opfylde regulatoriske krav.
Ved omhyggeligt at adressere hvert af disse trin, vil du samle de nødvendige beviser for at vise, at mediet opfylder de sikkerheds- og kvalitetsstandarder, der kræves til produktion af dyrket kød.
Hvad er de vigtigste forskelle mellem britiske og amerikanske regler for vækstmedier brugt i dyrket kød?
I Det Forenede Kongerige falder reguleringen af vækstmedier til dyrket kød under Novel Foods Regulation (EU Regulation 2015/2283), som er blevet bevaret i britisk lovgivning. Ethvert vækstmedie brugt i produkter, der ikke almindeligvis blev konsumeret før 15. maj 1997, skal gennemgå en formel vurdering af nye fødevarer af Food Standards Agency (FSA). Denne proces kræver indsendelse af detaljeret dokumentation, herunder information om mediets sammensætning, oprindelse og renhed. Derudover er en HACCP-baseret risikovurdering nødvendig for at demonstrere, hvordan forurenende stoffer kontrolleres under cellekulturprocessen.
Siden december 2025 har FSA implementeret en Cell-Cultivated Products sandbox. Dette initiativ tilbyder vejledning og understøtter hurtigere dataindsamling til ansøgninger om nye fødevarer.For at opnå endelig godkendelse skal virksomheder indsende et omfattende dossier, der adresserer mediesikkerhed, konsistens og validering af fremstilling. Først efter denne godkendelse kan produktet sælges i Storbritannien.
I modsætning hertil har USA ikke en specifik ny-mad-ramme skræddersyet til vækstmedier, hvilket gør direkte regulatoriske sammenligninger udfordrende. For virksomheder baseret i Storbritannien kan sourcing af mediekomponenter, der allerede overholder disse strenge standarder, forenkle godkendelsesprocessen.Hvordan understøtter Storbritanniens reguleringssandkasse validering af vækstmedier?
Storbritanniens reguleringssandkasse for dyrkede produkter giver en velorganiseret ramme, hvor virksomheder kan teste og forfine deres vækstmedieformuleringer. Overvåget af Food Standards Agency (FSA) og Food Standards Scotland (FSS), kører dette program i seks-måneders faser. I løbet af denne tid kan virksomheder udføre sikkerhedstests, foretage risikovurderinger og gennemgå dokumentation, mens de modtager værdifuld feedback fra regulatorer.
Denne praktiske tilgang tillader praktiske forsøg og trin-for-trin forbedringer, hvilket fremskynder indsamlingen af sikkerhedsdata og hjælper virksomheder med at tilpasse sig reguleringskrav. For dem, der arbejder med dyrket kød, kan sourcing af forhåndsgodkendte vækstmedier gennem









