

Si editas primero y verificas después, puedes corregir un cambio fuera de objetivo en el clon. Mantendría el flujo de trabajo simple: elige el método de edición de menor riesgo, mantén la exposición al editor corta y luego prueba tanto los sitios fuera de objetivo previstos como la estabilidad del clon antes de la liberación.
Para los ingenieros de bioprocesos, científicos de cultivo celular y equipos de I&D de carne cultivada, el punto principal es sencillo. Los sistemas CRISPR aún pueden cortar en sitios de coincidencia cercana, a menudo con 3–6 desajustes tolerados, y esos errores pueden trasladarse a clones de célula única expandidos. El artículo divide el control de riesgos en tres fases: antes de la edición, durante la edición, y después de la edición.
Aquí está la lista completa en términos sencillos:
-
Elija la herramienta de edición de menor riesgo para el trabajo
- Use edición base o edición prime cuando puedan realizar la edición sin una rotura de doble hebra
- Use modulación basada en dCas9 si solo necesita regulación génica
- Si necesita una nucleasa, comience con una variante de alta fidelidad Cas9
-
Asegure el material de partida
- Confirme la identidad de la línea celular
- Verifique micoplasma
- Registre el número de pasaje
- Secuencie el locus objetivo real en la línea de trabajo, no solo el genoma de referencia
-
Examine las guías antes del trabajo húmedo
- Utilice herramientas basadas en alineación y herramientas basadas en puntuación para off-target juntas
- Prefiera una guía con un perfil off-target más limpio sobre una con solo mayor actividad on-target
- Observe la longitud de la guía, 40–60% de contenido de GC, y carreras de homopolímeros
-
Limite la exposición dentro de la célula
- Utilice RNP o mRNA en lugar de sistemas plasmídicos o virales cuando sea posible
- Utilice la dosis mínima efectiva
- Evite extender la persistencia del editor solo para forzar resultados de transfección
-
Agregue controles adicionales para casos de mayor riesgo
- Considere nicasas emparejadas
- Utilice Cas9 escindido, inducible, o sistemas controlados por luz cuando el tiempo sea importante
- Agregue proteínas anti-CRISPR como un paso de apagado cuando sea necesario
-
Validar correctamente después de editar
- Confirmar la edición en el objetivo primero
- Verificar cada sitio predicho fuera del objetivo con NGS de amplicón dirigido
- Mover a GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, o WGS cuando el riesgo del proyecto es mayor
- Para editores de bases o editores primarios, agregar verificaciones a nivel de ARN donde sea relevante
-
No liberar un solo clon solo por la secuencia
- Comparar 2–3 clones independientes
- Usar un control parental no editado
- Eliminar clones con inestabilidad o deriva fenotípica
- Liberar solo cuando el estado de edición, la pantalla fuera del objetivo y los registros estén completos
Una forma breve de pensarlo: diseñar para evitar cortes fuera del objetivo, entregar para limitar el tiempo en la célula, luego validar a la profundidad que el riesgo del proyecto justifique. Ese es el hilo que recorre toda la pieza.
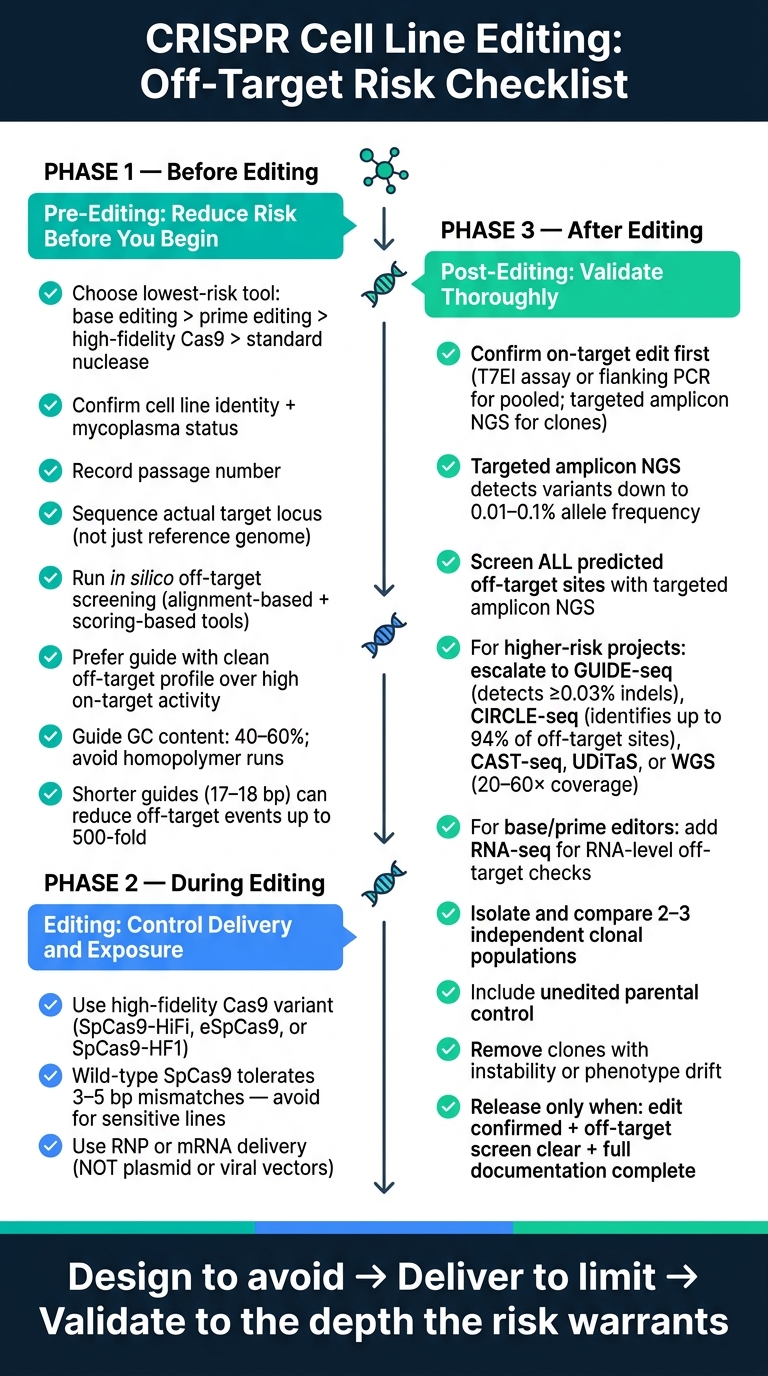
Control de Riesgo Fuera de Objetivo de CRISPR: Lista de Verificación en 3 Fases para la Edición de Líneas Celulares
Lista de verificación previa a la edición: reducir el riesgo antes de que comience la edición
Defina el objetivo de la edición y elija el método de edición de menor riesgo
Antes de ordenar un solo reactivo, tenga muy claro qué se pretende lograr con la edición. Un knockout, un knock-in, un cambio de un solo nucleótido y la modulación transcripcional no conllevan el mismo riesgo fuera de objetivo. Tampoco requieren la misma herramienta.
El orden general del riesgo es sencillo. Las nucleasas que forman DSB, como Cas9 y Cas12 , se sitúan en el extremo superior del riesgo porque pueden provocar grandes deleciones, translocaciones y respuestas al daño del ADN [1] [7]. Los editores de bases y los editores primarios utilizan nickases, por lo que evitan DSBs y reducen el riesgo de variación estructural [1][5]. Para la modulación transcripcional, los editores epigenéticos como dCas9 fusionados a modificadores transcripcionales dejan la secuencia de ADN sin cambios [1].
La regla práctica es simple: use el método menos genotóxico que aún pueda realizar la edición que necesita. Para cambios de un solo nucleótido, CBEs o ABEs son una mejor opción que HDR, que aún puede introducir indels [3] [1]. Para sustituciones y pequeñas inserciones o deleciones, la edición primaria a menudo muestra una menor actividad fuera del objetivo que el CRISPR-Cas9 estándar [1]. Si tienes que usar una nucleasa, elige una variante de alta fidelidad como SpCas9-HiFi, eSpCas9, o SpCas9-HF1 [1] [6].
Una vez que se establece el enfoque de edición, fija la línea celular de trabajo y la secuencia objetivo exacta.
Confirma la identidad de la línea celular, su historial y la secuencia del locus objetivo
Si la línea celular está mal identificada o contaminada, el resto del flujo de trabajo comienza a tambalearse. Incluso un ARN guía bien diseñado no rescatará un material de partida deficiente. Verifica la identidad de la línea celular antes de comenzar cualquier edición. Al mismo tiempo, confirma el estado de micoplasma y registra el número de pasaje actual, ya que las células de alto pasaje pueden alterar la estabilidad genómica y la eficiencia de edición [1][6].
Igual de importante, no te apoyes solo en un genoma de referencia.Secuenciar el locus objetivo exacto en la línea celular de trabajo. Ese paso te ayuda a identificar SNPs o indels que pueden bloquear la unión de la guía o crear nuevos sitios fuera del objetivo [1] [6].
Después de eso, pasa al diseño de la guía.
Realiza un cribado in silico de sitios fuera del objetivo antes de seleccionar reactivos
Una vez confirmado el locus objetivo, examina las guías de ARN candidatas in silico antes de comprometerte con el trabajo de laboratorio húmedo. Utiliza tanto herramientas basadas en alineación, como Cas-OFFinder o FlashFry, y herramientas basadas en puntuación, como la puntuación CFD o DeepCRISPR . El primer grupo ayuda a encontrar sitios genómicos con homología de secuencia. El segundo ayuda a clasificar esos sitios por probabilidad de corte predicha [1][5].
Cuando se están preseleccionando guías, un perfil más limpio fuera del objetivo debería superar la eficiencia bruta en el objetivo. Una guía con un 70% de eficiencia en el objetivo y sin predicciones de fuera del objetivo es un lugar más seguro para comenzar que una con un 90% de eficiencia y varios sitios de alto riesgo [6]. En algunos contextos, acortar la longitud de la guía de 20 pb a 17-18 pb puede reducir los eventos fuera del objetivo hasta 500 veces sin mucha pérdida de precisión en el objetivo [5]. Apunta a un contenido de GC entre el 40% y el 60%, y evita secuencias de cuatro o más bases idénticas [6][5].
Dicho esto, el cribado in silico tiene límites. No tiene en cuenta bien el estado de la cromatina, el ciclo celular o el contexto específico de la célula [1][6][4]. Piénsalo como un filtro, no como una prueba.Reduce el campo, pero no reemplaza la confirmación experimental.
Lleve los sitios predichos de mayor riesgo al plan de edición y validación.
sbb-itb-ffee270
Lista de verificación de edición: elección del editor de control, entrega y exposición
Utilice editores de alta especificidad y guías de ARN bien clasificadas
Comience con la lista corta de predicciones fuera del objetivo y úsela para elegir el editor. En la mayoría de los casos, una variante de alta fidelidad de SpCas9 - SpCas9-HiFi, eSpCas9 o SpCas9-HF1 - es una mejor opción por defecto que el SpCas9 de tipo salvaje [6] [1]. El SpCas9 de tipo salvaje puede tolerar hasta tres a cinco desajustes de pares de bases, especialmente en la región distal de PAM, y eso crea un riesgo significativo fuera del objetivo en líneas celulares sensibles [3].
Una regla simple ayuda aquí: usa el editor de alta fidelidad menos activo que aún entregue la edición deseada.
Para los editores base, rastrea ediciones colaterales y efectos fuera del objetivo en ARN por separado del riesgo fuera del objetivo en ADN [1] [8]. Esos son modos de falla diferentes y necesitan verificaciones separadas. Si puedes hacer la edición sin rupturas de doble cadena, la edición base o la edición primaria pueden encajar mejor en flujos de trabajo de mayor riesgo [1][8].
Una vez elegido el editor, el siguiente trabajo es mantener su tiempo dentro de la célula lo más corto posible.
Limita la persistencia del editor con entrega transitoria y dosis efectiva mínima
La persistencia del editor importa tanto como la elección del editor.Cuanto más tiempo el editor permanece activo en la célula, más tiempo tiene para actuar en sitios de baja probabilidad. Eso hace que el formato de entrega sea un punto de control importante.
Utilice entrega transitoria como RNPs o ARNm , y evite ADN plasmídico o vectores virales que extienden la expresión del editor [1] [5]. En la práctica, la entrega de RNP debería ser la opción predeterminada [6].
La dosis también importa. Una alta concentración de nucleasa aumenta la probabilidad de corte en sitios fuera del objetivo de baja sensibilidad [5]. Utilice la dosis mínima efectiva. Si la eficiencia de transfección es pobre, no simplemente añada más reactivo y espere lo mejor. Eso a menudo desplaza el problema en lugar de solucionarlo.
Agregar salvaguardas de precisión para flujos de trabajo de mayor riesgo
Algunos flujos de trabajo necesitan medidas de protección adicionales. Esto es especialmente cierto para objetivos cercanos a oncogenes, suppressors de tumores, o en líneas celulares sensibles a p53, donde un evento fuera del objetivo puede tener un costo desproporcionado [1][6][3].
Las salvaguardas útiles incluyen:
- Nickasas emparejadas, que requieren dos cortes cercanos. Un solo corte fuera del objetivo generalmente se repara sin mutación, por lo que el riesgo fuera del objetivo disminuye mucho en comparación con una configuración estándar de nucleasa [4][1].
- Sistemas Cas9 inducibles, controlados por luz o divididos, que ayudan a mantener la actividad del editor dentro de una ventana estrecha cuando la entrega es eficiente y la exposición debe ser breve [1].
- Proteínas Anti-CRISPR (Acr), que actúan como un interruptor de apagado. Estas proteínas Acr que ocurren naturalmente pueden desactivar el complejo CRISPR-Cas después de un intervalo definido, brindándole un freno molecular en la actividad del editor [1].
Lista de verificación post-edición: detectar eventos fuera del objetivo y validar clones
Examinar los sitios fuera del objetivo predichos con secuenciación dirigida
Una vez que se completa la edición, confirme primero el cambio previsto en el locus objetivo. Para una primera pasada rápida en células agrupadas, puede usar un ensayo de escisión por desajuste como T7 Endonuclease I, una digestión por restricción o una PCR flanqueante.Solo tenga cuidado con la interpretación: cada uno de estos métodos tiene límites de sensibilidad, especialmente para ediciones raras o variantes homocigotas [9].
Para la validación a nivel de clon, NGS de amplicón dirigido es el estándar. Le ofrece una vista cuantitativa de la frecuencia alélica y puede detectar variantes hasta 0.01% a 0.1% [3] .
Secuencie cada sitio predicho fuera del objetivo con NGS de amplicón dirigido. Ese debería ser el paso de validación por defecto.
Escale a ensayos a nivel del genoma o estructurales cuando el riesgo del proyecto sea mayor
La detección sitio por sitio no siempre es suficiente. Si el editor, el locus objetivo o la línea celular sugieren un riesgo oculto, pase a ensayos que puedan detectar eventos que no predijo de antemano.
Ensayos de descubrimiento a nivel del genoma como GUIDE-seq y CIRCLE-seq no necesitan listas previas de sitios fuera del objetivo.GUIDE-seq puede detectar sitios fuera del objetivo con frecuencias de indel tan bajas como 0.03% [2] . CIRCLE-seq puede identificar hasta 94% de los sitios fuera del objetivo in vitro [3] . Estos métodos son útiles cuando el contexto del tipo de célula puede enmascarar la actividad fuera del objetivo.
Si está preocupado por grandes reordenamientos, las lecturas estándar de amplicones pueden pasar por alto el problema principal. Las deleciones, inversiones y translocaciones necesitan ensayos diseñados para cambios estructurales, como CAST-seq y UDiTaS [1] .
La secuenciación del genoma completo (WGS) es la opción más amplia. Puede detectar indels, variaciones estructurales y cambios en el número de copias a lo largo del genoma [1]. El compromiso es profundidad y costo: generalmente necesita 20–60× cobertura, lo que lo hace poco adecuado para el cribado rutinario de poblaciones a granel [1].
Utilice NGS de amplicón dirigido para sitios predichos. Pase a ensayos a nivel de genoma o estructurales para proyectos de mayor riesgo. Para editores base o prime, agregue RNA-seq para verificar efectos fuera del objetivo a nivel de ARN.
Seleccione múltiples clones independientes y documente los criterios de liberación
Después de las verificaciones de secuencia, pruebe el fenotipo en más de un clon.
No avance con un solo clon editado. Aísle y expanda al menos dos a tres poblaciones clonales independientes y compárelas con un control parental no editado [4][9] . Elimine clones que muestren inestabilidad o deriva fenotípica [3]. Luego confirme la edición prevista en el estado de alelo requerido, ya sea heterocigoto o homocigoto, utilizando NGS de amplicón dirigido [9].
La documentación no es trabajo administrativo al final. Es parte de la liberación del clon. Registre el fondo de la línea parental, el diseño de sgRNA, la variante de nucleasa, el método de entrega y todos los resultados de QC [3]. Un clon solo debe avanzar cuando se confirme la edición prevista, los sitios predichos fuera del objetivo estén claros y el registro completo esté en su lugar.
Edición del Genoma con CRISPR: Cómo Minimizar Efectivamente los Efectos Fuera del Objetivo
Conclusión: una lista de verificación de tres fases para ediciones más limpias de líneas celulares
En conjunto, la lista de verificación trata el control fuera del objetivo como un proceso escalonado, no como una verificación de QC única. El objetivo es simple: reducir el riesgo temprano, limitar la actividad del editor y luego verificar el resultado.
La profundidad de validación debe coincidir con el riesgo. Libere solo múltiples clones independientes confirmados en el estado de alelo previsto.
Preguntas Frecuentes
¿Por qué no confiar en un solo clon?
Confiar en un solo clon es arriesgado. La edición CRISPR no es perfectamente específica, por lo que puede introducir mutaciones no deseadas fuera del objetivo.
Por eso los equipos suelen expandir múltiples poblaciones clonales. Hacer esto facilita encontrar una línea que lleve la edición en el objetivo deseado sin cambios perjudiciales fuera del objetivo.
Hay otra razón también: las líneas celulares pueden mostrar heterogeneidad genética. Secuenciar múltiples clones ayuda a confirmar que la eliminación homocigota u otra modificación del sitio objetivo está presente en todos los loci objetivo.
¿Cuándo es suficiente la secuenciación NGS de amplicones?
La secuenciación de próxima generación basada en amplicones es a menudo suficiente cuando necesitas una forma dirigida y rentable de confirmar posibles sitios fuera de objetivo señalados por herramientas computacionales u otros métodos de detección.
La secuenciación del genoma completo sigue siendo la única forma de cuantificar completamente los efectos fuera de objetivo. Pero para muchas aplicaciones, ese nivel de análisis simplemente no es necesario.
¿Cómo elijo el editor más seguro?
Elige la variante de nucleasa CRISPR menos activa que aún corte bien tu sitio en el objetivo.
No puedes elegir la mejor variante solo con la predicción. La única forma confiable es realizar una pequeña prueba en variantes de nucleasas seleccionadas y leer la edición con secuenciación de próxima generación.
Para la I&D de carne cultivada, eso te da un camino práctico hacia adelante: comienza con una lista corta de variantes, luego prueba las más débiles paso a paso hasta que encuentres la opción menos activa que aún edita el sitio objetivo de manera eficiente.









