

En salas limpias, el cumplimiento de GMP garantiza calidad y seguridad consistentes al requerir un monitoreo detallado y registros de datos precisos. Para las instalaciones de carne cultivada, esto es particularmente importante, ya que incluso desviaciones menores en las condiciones de la sala limpia pueden comprometer el crecimiento celular o contaminar lotes de producción.
Puntos clave:
- Normas GMP: Enfocarse en la integridad de los datos, siguiendo el marco ALCOA+ (Atribuible, Legible, Contemporáneo, Original, Preciso, Completo, Consistente, Duradero, Disponible).
- Parámetros Críticos: Monitorear partículas en el aire, conteos microbianos, temperatura, humedad y presión para detectar riesgos temprano. Esto requiere seleccionar sensores precisos capaces de mantener estos parámetros críticos.
- Sistemas de Datos: Usar sistemas de control de bioprocesos validados con acceso basado en roles, registros de auditoría y almacenamiento seguro para registros electrónicos y en papel.
- Riesgos Comunes: Evite errores en el manejo manual de datos, cambios de configuración sin control y prácticas de almacenamiento inadecuadas.
- GMP Personalizado para Carne Cultivada: Ajuste las estrategias de monitoreo para abordar riesgos únicos como las condiciones del biorreactor y los residuos de agentes de limpieza.
Para I&D de carne cultivada, una gestión de datos robusta garantiza la seguridad del producto, el cumplimiento normativo y operaciones escalables. Aborde proactivamente las vulnerabilidades conocidas para evitar problemas regulatorios costosos más adelante.
Requisitos Clave de GMP para la Integridad de Datos en Salas Limpias
Comprensión de los Principios ALCOA+
La piedra angular de la integridad de datos GMP reside en el marco ALCOA+. Organismos reguladores como el MHRA, EMA, y OMS lo utilizan para determinar si se puede confiar en los registros de salas limpias.ALCOA+ significa: Atribuible, Legible, Contemporáneo, Original, Preciso, Completo, Consistente, Duradero, y Disponible. Cada uno de estos términos tiene importancia práctica en las operaciones de salas limpias.
- Atribuible: Cada entrada - ya sea un conteo de partículas, lectura de presión o registro de limpieza - debe mostrar claramente quién la registró, junto con la fecha, hora y detalles relevantes del instrumento.
- Legible: Los registros deben ser fáciles de leer y descifrar, asegurando claridad durante las revisiones o inspecciones.
- Contemporáneo: Los datos deben registrarse en tiempo real. Las entradas retrasadas o retrospectivas pueden comprometer la fiabilidad de los registros.
- Original: Los datos deben permanecer en su forma capturada inicialmente, sin ediciones o alteraciones no autorizadas.
- Preciso: Los valores registrados deben reflejar verdaderamente los resultados observados, libres de errores o manipulaciones.
- Completo: Todos los registros relevantes, incluidas las desviaciones o resultados fuera de especificación, deben ser documentados.
- Consistente, Duradero, y Disponible: Los registros deben seguir la secuencia correcta, preservarse intactos durante el período de retención requerido y estar fácilmente accesibles para revisión o inspección.
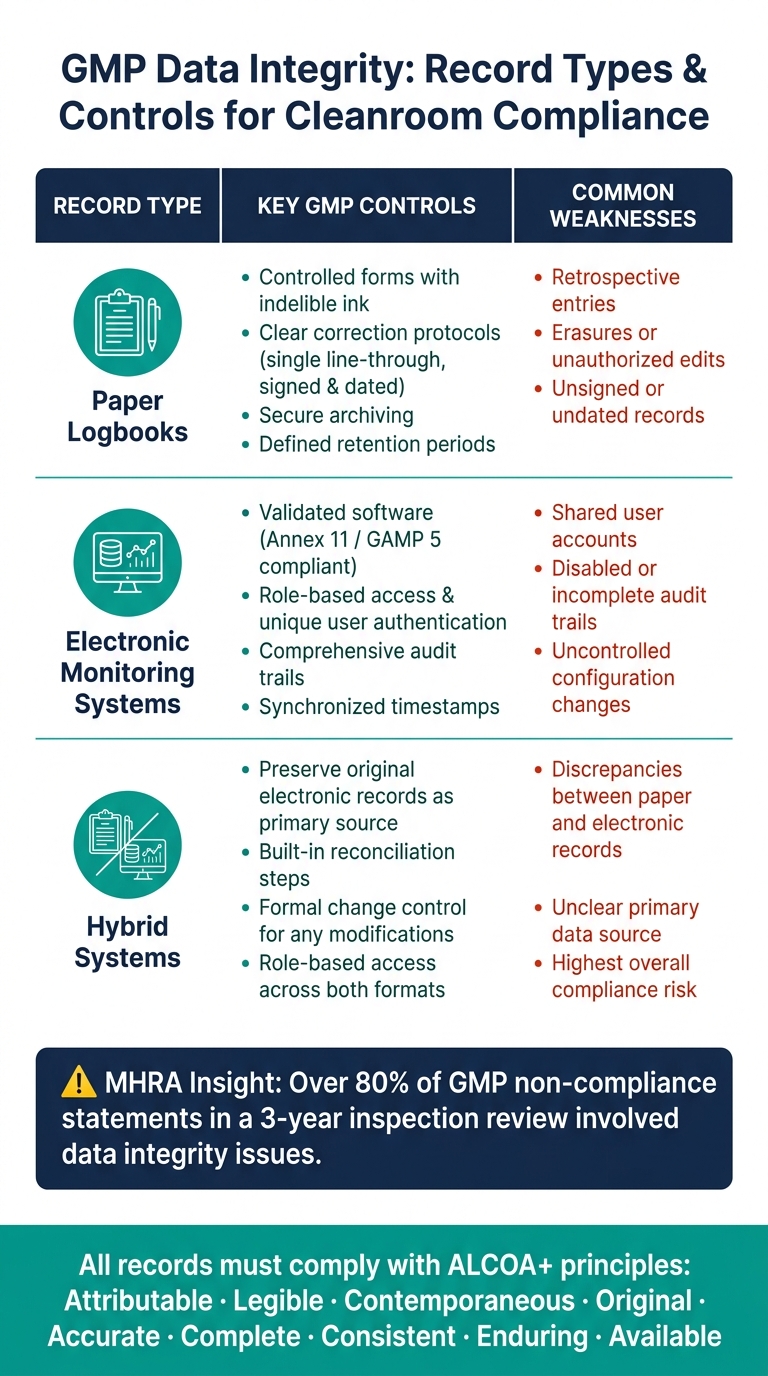
Los reguladores ponen un fuerte énfasis en estos principios. Por ejemplo, una revisión de inspección de MHRA reveló que más del 80% de las declaraciones de incumplimiento de GMP en un período de tres años involucraron problemas de integridad de datos [5]. Para integrar ALCOA+ en los flujos de trabajo diarios, las instalaciones pueden adoptar formularios bien estructurados, imponer campos obligatorios y realizar revisiones regulares de la pista de auditoría.
Con ALCOA+ como base, el siguiente paso es asegurar que estos principios se mantengan en sistemas de papel, electrónicos e híbridos.
Asegurando la Integridad de los Datos a Través de Formatos
En las instalaciones de carne cultivada, donde los datos influyen directamente en las decisiones de liberación de lotes, mantener la integridad en todos los formatos de registro es innegociable. Las GMP requieren el mismo nivel de integridad tanto para los registros en papel como para los electrónicos, aunque los controles específicos pueden diferir según el formato.
- Sistemas de papel: Las mejores prácticas incluyen el uso de formularios controlados con tinta indeleble y protocolos claros de corrección (e.g. , correcciones con una sola línea tachada, firmas y fechas). El archivo seguro y la adherencia a los períodos de retención definidos también son críticos.
- Sistemas electrónicos: Estos deben operar en software validado que cumpla con Annex 11 y GAMP 5 . Características clave incluyen acceso basado en roles, autenticación única de usuarios, registros de auditoría completos y marcas de tiempo sincronizadas. Las revisiones regulares de los registros de auditoría son esenciales para identificar y abordar cualquier irregularidad.
- Sistemas híbridos: Estos representan el mayor riesgo ya que involucran tanto registros electrónicos como en papel. Por ejemplo, cuando un instrumento genera datos electrónicos que luego se transcriben en un registro en papel, la salida electrónica original debe preservarse como el registro principal. Los pasos de reconciliación deben integrarse en el flujo de trabajo para detectar y resolver cualquier discrepancia entre los registros electrónicos y en papel. Esto es particularmente crítico en la producción de carne cultivada, donde incluso pequeñas inconsistencias de datos podrían comprometer las medidas de control de contaminación.
La tabla a continuación resume los controles clave y las debilidades comunes para cada tipo de registro:
| Tipo de Registro | Controles GMP Clave | Debilidades Comunes |
|---|---|---|
| Formularios controlados, tinta indeleble, protocolos claros de corrección, entradas firmadas y fechadas | Entradas retrospectivas, borraduras, registros sin firmar | |
| Sistemas de monitoreo electrónico | Software validado, acceso basado en roles, registros de auditoría, sincronización de tiempo | Cuentas de usuario compartidas, registros de auditoría deshabilitados o incompletos |
| Sistemas híbridos | Preservar los registros electrónicos originales; implementar pasos de reconciliación | Discrepancias entre registros en papel y electrónicos, fuente de datos primaria poco clara |
Para garantizar el cumplimiento, los registros deben ser categorizados según su criticidad.Para las instalaciones de carne cultivada, los datos relacionados con las decisiones de liberación de lotes o el control de contaminación (e.g. , resultados de monitoreo ambiental, registros de alarmas de HVAC, o datos de pruebas de integridad de filtros) deben estar sujetos a los controles de acceso más estrictos, revisiones frecuentes y una gestión robusta de la trazabilidad de auditoría.
sbb-itb-ffee270
Monitoreo Ambiental de Rutina en Salas Limpias GMP& Integridad de Datos 21CFR parte 11
Gestión del Ciclo de Vida de Datos en Salas Limpias
Integridad de Datos GMP: Tipos de Registros& Controles para el Cumplimiento en Salas Limpias
Etapas del Ciclo de Vida de Datos en Salas Limpias
El ciclo de vida de datos en salas limpias en instalaciones de carne cultivada involucra varias etapas, cada una con requisitos específicos de cumplimiento.
La generación de datos marca el comienzo. Esto incluye lecturas de instrumentos como contadores de partículas, sensores de presión diferencial, sondas de temperatura y humedad, muestreadores de aire viables, placas de contacto de superficie y registros de verificación de limpieza. Para cada parámetro, debe haber una frecuencia de muestreo documentada, un operador designado y un instrumento calibrado. Alinear estas tareas de monitoreo con las etapas de producción, como la inoculación, la expansión celular o la cosecha, ayuda a demostrar cómo el control ambiental se vincula directamente con la calidad y seguridad del producto.
Una vez generados, los datos ingresan a la etapa de captura y transferencia. Idealmente, los sistemas electrónicos deben registrar automáticamente las lecturas con entradas con marca de tiempo vinculadas a cuentas de usuario individuales. Para las entradas en papel, los datos deben registrarse en tiempo real utilizando tinta indeleble, con verificaciones de conciliación al transferir los datos a sistemas electrónicos.
La fase de almacenamiento es igualmente crítica.Tanto los datos en bruto como los procesados deben preservarse para que cualquier valor reportado pueda rastrearse hasta su registro original. Esto requiere repositorios seguros y validados con controles de acceso basados en roles y pruebas regulares de respaldo. Las copias de seguridad deben almacenarse en una ubicación separada del sistema principal y verificarse periódicamente para garantizar que puedan restaurarse cuando sea necesario.
Finalmente, el archivado concluye el ciclo de vida. Los registros pasan a un estado de solo lectura y acceso controlado una vez que ya no se utilizan activamente, pero deben permanecer recuperables durante el período de retención requerido. En las instalaciones de carne cultivada, el archivado de datos de la fase de desarrollo también puede apoyar los esfuerzos de validación futuros.
Una comprensión clara de estas etapas es esencial para gestionar los riesgos de manera efectiva, como se describe a continuación.
Riesgos Comunes en la Gestión de Datos
El manejo de datos durante las transferencias plantea riesgos considerables.Los errores de transcripción manual y las entradas retrospectivas pueden socavar la integridad de los datos. Para evitar esto, todas las entradas deben adherirse a los principios ALCOA+ (Atribuible, Legible, Contemporáneo, Original, Preciso, más Completitud, Consistencia, Duradero y Disponible) en tiempo real.
Los cambios de configuración son otra preocupación importante. Los ajustes en los límites de alarma, los mapeos de sensores o la configuración del reloj del sistema sin un control de cambios formal pueden comprometer la fiabilidad de los datos registrados antes y después del cambio. Además, las fallas de almacenamiento - ya sea debido a bases de datos corruptas, copias de seguridad no probadas o archivos en papel dañados por factores ambientales - pueden hacer que los registros críticos sean inaccesibles. Para mitigar estos riesgos, asegúrese de que cada flujo de datos esté mapeado a un punto de archivo designado con una propiedad clara, reduciendo la probabilidad de que se señalen vulnerabilidades durante las inspecciones regulatorias.
Controles GMP para Sistemas de Monitoreo de Salas Limpias
Controles Críticos para Sistemas de Monitoreo
Un sistema de monitoreo que cumple con los estándares GMP se basa en sensores calibrados, manejo seguro de datos y gestión efectiva de alarmas. Los sensores para parámetros como temperatura, humedad relativa, presión diferencial, conteo de partículas no viables y muestreo microbiano viable deben calibrarse según cronogramas documentados y ser trazables a estándares reconocidos. Automatizar la transferencia de datos desde estos sensores, con marcas de tiempo sincronizadas, minimiza el riesgo de errores manuales.
La gestión de alarmas es igualmente crucial. Los límites de alarma deben alinearse con marcos regulatorios como los límites de clase de ISO 14644-1 y la guía de EU GMP Anexo 1. Cada alarma activada debe ir acompañada de una respuesta registrada, incluyendo detalles del usuario, marcas de tiempo y cualquier comentario.No documentar una respuesta a una alarma crea una vulnerabilidad de cumplimiento.
Los controles de acceso basados en roles deben ser estrictamente aplicados en todo el sistema. Se debe requerir autorización a nivel de administrador para cualquier cambio en los límites de alarma, configuraciones de sensores o ajustes del reloj del sistema, y estos cambios deben seguir un proceso formal de control de cambios. Los registros de auditoría son obligatorios para todas las acciones relacionadas con GMP, como actualizaciones de configuración, eliminaciones de datos, firmas electrónicas y ajustes de sensores. Estos registros necesitan revisiones regulares, como se describe en la guía de integridad de datos de MHRA y el Anexo 11 de GMP de la UE.
Para los sistemas de producción de carne cultivada, estos controles son particularmente importantes, ya que las condiciones ambientales tienen un impacto directo en la viabilidad celular.
Una vez que estos controles están en su lugar, el sistema debe ser validado y los cambios gestionados cuidadosamente para asegurar el cumplimiento continuo.
Procedimientos de Validación y Control de Cambios
Los sistemas de monitoreo deben someterse a validación a través de las etapas IQ, OQ y PQ para verificar la precisión de los sensores, la funcionalidad de las alarmas, la integridad de los datos, los procesos de respaldo y las pistas de auditoría. Alternativamente, se puede utilizar un enfoque de ciclo de vida alineado con los principios de GAMP 5 y el Anexo 11.
El Anexo 1 de las GMP de la UE (revisión de 2022) requiere que los sistemas de monitoreo ambiental estén "adecuadamente calificados y validados" y exige que los registros electrónicos cumplan con los estándares del Anexo 11 para integridad, seguridad y trazabilidad. Estos requisitos establecen la base para cualquier instalación conforme a GMP.
Cualquier modificación que pueda afectar la integridad de los datos, la funcionalidad de las alarmas o la trazabilidad debe pasar por un proceso formal de control de cambios. Incluso actualizaciones aparentemente menores, como parches de software, pueden interrumpir las pistas de auditoría y no deben implementarse sin una evaluación de impacto previa.
Diferentes tipos de datos requieren controles GMP adaptados para garantizar informes precisos y oportunos.
Comparación de Tipos de Datos y Requisitos de Cumplimiento
Cada tipo de dato en el monitoreo de salas limpias tiene requisitos específicos para mantener el cumplimiento de GMP.La tabla a continuación describe los controles clave para varios tipos de datos:
| Tipo de datos | Modo de monitoreo | Controles clave de GMP | Base de límites |
|---|---|---|---|
| Conteo de partículas no viables | Continuo o frecuente durante operaciones en Grado A/B; rutinario en otros grados | Contadores de partículas validados; captura automática de datos; con alarma; calibración trazable a estándares; registro de auditoría para cambios de configuración | Límites de clase ISO 14644-1; guía del Anexo 1 para zonas de Grado A/B |
| Presión diferencial | Continuo; con alarma | Transmisores de presión calibrados; grabación automática con marca de tiempo; registros de reconocimiento de alarmas; mantener diferencial de 10–15 Pa entre zonas | Anexo 1; diseño de clasificación de salas específico de la instalación |
| Temperatura y humedad relativa | Continuo para procesos críticos; periódico en otros lugares | Sondas calibradas; captura automática de datos; análisis de tendencias; límites de alarma basados en necesidades del proceso y regulatorias | Conocimiento del proceso; orientación regulatoria; sensibilidad del producto |
| Microbios viables en el aire | Intermitente (muestreo de aire activo); frecuencia aumentada para operaciones críticas | Muestreadores calificados; procedimientos de muestreo controlados; cadena de custodia al laboratorio; resultados vinculados a lote y ubicación; registros listos para investigación | Límites microbianos del Anexo 1 de EU GMP por grado |
| Resultados de contacto superficial | Periódico; post-limpieza y post-operación | Métodos de muestreo controlados; trazabilidad de laboratorio; resultados revisados contra límites específicos de grado; vinculados a registros de limpieza | Anexo 1 de las GMP de la UE; SOPs de la instalación |
Cada tipo de dato requiere criterios de aceptación definidos, horarios de revisión regular, políticas de retención y un proceso de escalada para desviaciones.Aplicar estándares de revisión uniformes a todos los tipos de datos es un error común que los reguladores están examinando cada vez más. Adaptar el proceso de revisión a las necesidades específicas de cada tipo de dato garantiza el cumplimiento y la eficiencia operativa.
Informes, Revisión y Acciones Correctivas
Creación de Informes Cumplidores
Los informes alineados con los estándares ALCOA+ deben ser exhaustivos, precisos y accesibles para auditorías. Un informe de monitoreo de sala limpia conforme a GMP debe ser conciso, verificable y capaz de respaldar decisiones de liberación de lotes mientras demuestra control ambiental. Como mínimo, estos informes deben:
- Cubrir el período y alcance del monitoreo.
- Resumir las actividades de muestreo en comparación con el cronograma planificado.
- Indicar claramente si se excedieron los límites de alerta o acción.
El análisis de tendencias es un componente clave de estos informes, empleando herramientas estadísticas como gráficos de control, promedios móviles y tasas de excursión por cada 100 muestras para identificar cambios graduales. Por ejemplo, una tendencia mensual que muestra un aumento constante en los recuentos viables cerca de una línea de cosecha de biorreactores proporciona mucho más conocimiento que un solo evento fuera de los límites. Agregar anotaciones, como actividades de mantenimiento, ajustes de procesos o cambios de personal, hace que los datos sean más fáciles de interpretar y estén más listos para auditorías.
Las revisiones de la pista de auditoría son otro paso crítico, que requiere personal capacitado para documentar meticulosamente sus hallazgos. Esto incluye registrar quién revisó eventos específicos del sistema, anotar cualquier anomalía y detallar acciones de seguimiento, todo dentro de un registro firmado y fechado.
La frecuencia de los informes debe alinearse con el riesgo asociado. Por ejemplo:
- Los informes asociados a lotes se crean para cada ejecución de producción.
- Los resúmenes de monitoreo ambiental de rutina son típicamente semanales o mensuales.
- Los informes de tendencias se preparan mensualmente o trimestralmente para identificar signos tempranos de desviación.
La frecuencia de informes elegida debe justificarse en los procedimientos operativos estándar (SOPs) y seguirse de manera consistente. Estos protocolos también proporcionan la base para iniciar acciones correctivas cuando se identifican desviaciones.
Abordar Fallos de Cumplimiento
Cuando ocurren desviaciones, una respuesta estructurada y rastreable es esencial. Cada desviación debe tener un identificador único, una descripción clara y una evaluación de riesgos que evalúe tanto el impacto en el producto como la integridad de los datos. Las desviaciones también deben clasificarse (menor, mayor o crítica) y vincularse a lotes específicos o corridas de producción para evaluar si se ve afectada la liberación del lote o si se necesita realizar pruebas adicionales.
El marco CAPA (Acción Correctiva y Preventiva) es fundamental para abordar fallos en las GMP. Un CAPA efectivo requiere más que atribuir eventos a "error humano". La guía de EMA y PIC/S enfatiza:
"la falta de investigación adecuada de desviaciones críticas, resultados OOS y problemas de integridad de datos" es una causa recurrente de acciones de cumplimiento.
Las herramientas de análisis de causa raíz, como los 5 Porqués o los diagramas de espina de pescado, son invaluables para descubrir problemas sistémicos, ya sea que se relacionen con brechas en los procedimientos, capacitación insuficiente o debilidades en los controles técnicos. Las acciones correctivas deben abordar los riesgos identificados durante la captura y almacenamiento de datos.
Cada CAPA debe incluir criterios de efectividad medibles. Por ejemplo: "no se repiten excursiones viables de Grado B por encima del nivel de acción durante seis meses". Además, las revisiones de seguimiento son esenciales para asegurar que se cumplan estos criterios.Métricas comunes de CAPA incluyen:
- El número de acciones abiertas.
- Tiempo promedio para el cierre.
- Porcentaje de acciones completadas a tiempo.
- Tasa de desviaciones repetidas, que sirve como un fuerte indicador de la efectividad de CAPA.
Una revisión de las cartas de advertencia de GMP de 2015 a 2019 reveló que el 65–70% de las citaciones de integridad de datos se debieron a investigaciones inadecuadas, documentación faltante o fallas en la revisión y reporte de datos correctamente [2]. Esto resalta la importancia de un reporte robusto y un marco CAPA receptivo como evidencia de una instalación bien controlada.
Mantener el Cumplimiento de GMP en Instalaciones de Carne Cultivada
Para garantizar la seguridad y calidad en la producción de carne cultivada, las instalaciones deben adaptar los controles de GMP establecidos para enfrentar los desafíos específicos de este campo emergente.Dado que la integridad de los datos de la sala limpia juega un papel crítico en el mantenimiento de la seguridad del producto, es esencial refinar las prácticas de GMP para la carne cultivada.
Adaptación de las Prácticas de GMP para la Carne Cultivada
Los marcos de GMP como el Anexo 1 de la UE, originalmente creados para productos farmacéuticos, requieren ajustes para abordar los riesgos únicos en la producción de carne cultivada. Una evaluación formal de riesgos, como un análisis al estilo FMEA o HACCP, proporciona una base sólida para alinear los principios de GMP con cada etapa de la producción. Operaciones críticas como la descongelación del banco de células, la inoculación del biorreactor, la expansión celular y la cosecha demandan clasificaciones de sala limpia apropiadas, protocolos de vestimenta y monitoreo ambiental según lo especificado en el Anexo 1.Mientras tanto, las tareas posteriores como el manejo de andamios y el empaquetado pueden adherirse a los estándares de higiene de grado alimenticio GMP bajo Reglamento (CE) No 852/2004, siempre que la trazabilidad y la integridad de los datos se mantengan intactas durante todo el proceso [6] [9][14].
Las estrategias de monitoreo ambiental deben centrarse en organismos relevantes para la carne cultivada y la seguridad alimentaria, en lugar de enfocarse únicamente en patógenos farmacéuticos tradicionales. El muestreo debe priorizarse en áreas de alto riesgo, como aquellas cerca de biorreactores abiertos, zonas de preparación de medios y estaciones de manejo de andamios. Estas ubicaciones deben seleccionarse en base a patrones documentados de flujo de aire y análisis de movimiento del personal [9][10].
Dado el alto volumen de datos generados por los biorreactores de carne cultivada, los sistemas deben ser capaces de capturar, marcar con fecha y hora, y almacenar de manera segura estos datos en un repositorio validado. El archivo de datos crudos original del instrumento siempre debe identificarse como el registro principal para garantizar el cumplimiento [7][8].
Los protocolos de limpieza y desinfección también requieren una consideración cuidadosa. Los residuos considerados aceptables en entornos farmacéuticos podrían interferir con la adhesión o diferenciación celular en la producción de carne cultivada. Los datos de verificación para los agentes de limpieza deben recopilarse y mantenerse como parte del programa de monitoreo ambiental [3][4].
Utilizando recursos de la industria como Cellbase
Las plataformas de adquisición especializadas son invaluables para satisfacer las necesidades específicas de las instalaciones de carne cultivada. El equipo de sala limpia compatible con GMP debe cumplir con los estándares de protección contra ingreso y limpieza, al tiempo que se integra perfectamente con los sistemas de datos validados. Los proveedores deben proporcionar especificaciones detalladas, incluyendo capacidades de seguimiento de auditoría, formatos de exportación de datos, configuraciones de alarmas y procedimientos de calibración, junto con el equipo.
Al obtener sistemas a través de
- Características de integridad de datos: auditorías seguras, registros con marca de tiempo, permisos basados en roles y inicios de sesión de usuario únicos
- Compatibilidad del sistema: soporte para protocolos de comunicación estándar y APIs para almacenamiento centralizado de datos
- Calibración y mantenimiento: disponibilidad de documentación completa
- Soporte de calificación: plantillas IQ/OQ proporcionadas por el proveedor para una validación simplificada
- Adecuación para salas limpias: materiales y diseños que faciliten la limpieza
Solicitar la documentación del proveedor al principio del proceso de adquisición puede ayudar a evitar posibles problemas de calificación en el futuro [3][4].
Conclusión
Resumen de Puntos Clave
El cumplimiento de GMP en la gestión de datos de salas limpias gira en torno a demostrar control: sobre procesos, registros y las decisiones informadas por esos datos. Si un registro carece de fiabilidad, el proceso que documenta se vuelve igualmente cuestionable. Este principio se aplica universalmente, ya sea tratando con registros de monitoreo ambiental, salidas de biorreactores, informes de desviación o certificados de calibración.
Cuatro temas centrales han surgido a lo largo de esta discusión. Primero, la integridad de los datos, guiada por los principios ALCOA+, es la piedra angular de la documentación de salas limpias conforme. Segundo, la gestión del ciclo de vida asegura que los datos se registren con precisión, se revisen puntualmente, se almacenen de manera segura y se conserven durante el tiempo requerido. Tercero, los sistemas de monitoreo validados y controlados por cambios forman la base técnica que ningún SOP puede reemplazar.Como se destacó en el análisis de las inspecciones de GMP de la MHRA de 2016 a 2021, las deficiencias comunes continúan incluyendo registros incompletos y revisiones insuficientes de la trazabilidad de auditoría [1]. Finalmente, informes precisos y trazables aseguran que los datos brutos puedan vincularse a decisiones de lote, investigaciones y acciones correctivas, cumpliendo con las expectativas regulatorias.
Para las instalaciones de carne cultivada, estos principios adquieren aún mayor importancia. El desafío de combinar flujos de trabajo estilo I&D con controles a nivel de producción exige una gobernanza de datos robusta para unir ambos entornos operativos. La gestión adecuada de datos en salas limpias no solo asegura consistencia y reproducibilidad, sino que también prepara las instalaciones para la ampliación mientras demuestra la seguridad del producto a reguladores, inversores y consumidores.
¿El consejo más práctico? Abordar los riesgos conocidos antes de que los auditores los destaquen.Las vulnerabilidades como los sistemas híbridos de papel y electrónico, los inicios de sesión compartidos de usuarios, las revisiones de datos retrasadas y el almacenamiento local no controlado son previsibles y prevenibles. Resolver proactivamente estos problemas es mucho más efectivo - y menos costoso - que reconstruir un rastro de datos después de un incidente de calidad.
Para los equipos que buscan equipos de monitoreo, sensores o infraestructura adaptada a estas necesidades,
Preguntas Frecuentes
¿Cómo se puede demostrar ALCOA+ en los registros diarios de salas limpias?
Para aplicar los principios de ALCOA+ en los registros diarios de salas limpias, asegúrese de lo siguiente:
- Atribuible: Identifique claramente a la persona responsable, incluyendo marcas de tiempo para cada entrada.
- Legible : Los registros deben ser fáciles de leer y estar libres de ambigüedades.
- Contemporáneo: Documentar la información en el momento en que ocurre la actividad.
- Original: Conservar la primera grabación de datos, no copias o transcripciones.
- Preciso: Asegúrese de que todas las entradas reflejen los datos verdaderos sin errores.
- Completo: Incluir todos los datos y metadatos relevantes sin omisiones.
- Consistente: Mantener un orden lógico y secuencial en los registros.
- Duradero: Utilizar formatos y materiales adecuados para la preservación a largo plazo.
- Disponible: Mantener los registros accesibles para revisión o auditorías cuando sea necesario.
Estos pasos son críticos para asegurar el cumplimiento de Good Manufacturing Practice (GMP) en la gestión de datos de salas limpias.
¿Cuáles son los principales riesgos de integridad de datos en sistemas híbridos de papel-electrónico?
Almacenar información en múltiples ubicaciones introduce complicaciones en la verificación de la precisión de los datos. Además, la entrada manual de datos aumenta el riesgo de errores humanos, mientras que los sistemas mal controlados o independientes dejan los registros vulnerables a la manipulación o eliminación. Estos problemas subrayan la necesidad de prácticas sólidas de gestión de datos para mantener el cumplimiento y preservar la integridad de los datos.
¿Qué evidencia esperan los inspectores para la validación del sistema y el control de cambios?
Los inspectores a menudo solicitan evidencia documentada que demuestre la validación del sistema. Esto incluye probar parámetros críticos como:
- Integridad del filtro HEPA: Asegurando que los filtros cumplan con los estándares de rendimiento requeridos.
- Diferenciales de flujo de aire y presión: Verificando que estos estén dentro de rangos aceptables para mantener ambientes controlados.
- Datos de monitoreo ambiental: Demostrando que la instalación cumple con los requisitos de limpieza y control de contaminación.
Más allá de las pruebas de validación, mantener registros de actividades de control de cambios es igualmente importante. Esto cubre acciones como reemplazos de filtros o modificaciones de instalaciones, que ayudan a demostrar que el sistema sigue funcionando como se espera y cumple con los estándares regulatorios.










