

Erätiedot ovat kriittisiä vaatimustenmukaisuuden ja tuoteturvallisuuden kannalta. Ne dokumentoivat jokaisen tuotantovaiheen varmistaen, että sääntelyvaatimukset täyttyvät. Viljellyn lihan tuottajille steriiliyden ylläpitäminen ja yksityiskohtaiset tiedot ovat ehdottomia. FDA-tarkastukset korostavat usein ongelmia, kuten puuttuvia tietoja, keskeneräisiä tarkastuksia ja huonoja korjaavia toimenpiteitä, jotka voivat johtaa varoituksiin tai häiriöihin.
Keskeiset huomiot:
- Erätiedot: Kaksi tyyppiä - Master Batch Record (MBR) (resepti) ja Batch Production Record (BPR) (suoritus).
- Yleiset ongelmat: Inhimilliset virheet ( 50% ongelmista), puuttuvat prosessin aikaiset tarkastukset, keskeneräiset tarkastukset ja huonot CAPA (Korjaavat ja ennaltaehkäisevät toimenpiteet) -järjestelmät.
- FDA-standardit: ALCOA+ -periaatteiden (Attribuoitava, Luettava, Ajankohtainen, Alkuperäinen, Tarkka, Täydellinen, Johdonmukainen, Kestävä, Saatavilla) noudattaminen on pakollista.
- Ratkaisut: Riippumattomat tarkastukset, sähköiset eräkirjat ja tiukka toimittajien tarkistus voivat minimoida virheitä ja parantaa vaatimustenmukaisuutta.
Viljellyn lihan yritykset, kuten UPSIDE Foods, ovat asettaneet vertailukohdan varmistamalla yksityiskohtaisen dokumentoinnin, syötteiden jäljitettävyyden ja nopeat korjaavat toimenpiteet. Oppimalla näistä käytännöistä tuottajat voivat välttää sääntelyyn liittyvät sudenkuopat ja ylläpitää korkealaatuisia standardeja.
Kattava opas dokumentointiin ja kirjanpitoon FDA-vaatimustenmukaisuudelle biotieteissä
sbb-itb-ffee270
Yleiset ongelmat eräkirjojen dokumentoinnissa
FDA:n tarkastusraportit korostavat johdonmukaisesti toistuvaa ongelmaa: tuotantotietojen tarkastuksen poikkeamat ovat yksi yleisimmistä GMP-puutteista, joita sääntelyviranomaiset mainitsevat [7]. Viljellyn lihan tuottajille nämä puutteet menevät pelkkien hallinnollisten virheiden ulkopuolelle - ne vaarantavat kyvyn osoittaa jatkuvat steriilit olosuhteet. Nämä ongelmat ilmenevät useissa muodoissa, kuten alla olevissa esimerkeissä on kuvattu.
Keskeneräiset Arviot ja Poikkeamat
Yksi yleinen ongelma on, että laadunvalvontayksiköt eivät tarkasta eräkirjoja perusteellisesti. Sen sijaan, että tarkastukset olisivat olennainen osa vapautusprosessia, ne tapahtuvat usein reaktiivisesti - vasta kun tuoteongelma on jo ilmennyt [7]. Tämä lähestymistapa jättää merkittäviä aukkoja tuotantokirjoihin.
Esimerkiksi, Davis City Pharmacy sai FDA 483 -huomautuksen, koska eräkirjoista puuttui kriittisiä tietoja, kuten komponenttimäärät, toimintavaiheet ja henkilöstön nimikirjaimet. Samoin, CAPS sai huomautuksen puuttuvista vaadituista allekirjoituksista ja tarkastajan varmennuksista keskeisissä merkinnöissä [3]. Nämä puutteet eivät ole yksittäisiä tapauksia; tutkimukset osoittavat, että noin 52% dokumentointirikkomukset lisääntyvät, kun vankat bioprosessinhallintajärjestelmät puuttuvat [3].
"Ei ole prosessin monimutkaisuus, joka aiheuttaa huomautuksia - vaan epäjohdonmukaisuus, epätäydellisyys ja huono valvonta." - GXP Auditing & Consulting Services [5]
Puutteelliset prosessintarkastustiedot
Toinen yleinen puute on asianmukaisen dokumentoinnin puuttuminen prosessintarkastuksista. Nämä tiedot ovat ratkaisevan tärkeitä, erityisesti kriittisissä valvontapisteissä aseptisissa toiminnoissa. Esimerkiksi Nephron Sterile Compounding Center sai huomautuksia, koska se ei dokumentoinut olennaisia pukeutumisvaiheita ja aseptisia menettelyjä eräkirjoissaan [3]. Viljellyn lihan tuottajille, joille steriiliys on ensiarvoisen tärkeää, tällaiset laiminlyönnit tekevät mahdottomaksi varmistaa kontaminaation hallintatoimenpiteiden.
noudattamisen.Amphastarille annettiin myös huomautus siitä, että se ei tutkinut tai dokumentoinut odottamattomia saantovaihteluita tai tuotantoeroja [3]. Tällaisten laiminlyöntien riskit ovat ilmeisiä. Eräässä tapauksessa tunnistamaton lääkelaitos vuosina 2024/2025 havaittiin säilyttävän valmiita erärekistereitä avoimilla hyllyillä ja pöydillä. FDA:n tutkijat löysivät puuttuvia sivuja, mukaan lukien seitsemän yhdestä rekisteristä, ja koko "Synthesis Solution" -osio puuttui toisesta [6].
CAPA ja toimittajan GMP:n varmennuksen epäonnistumiset
Dokumentointivirheiden lisäksi tehokkaiden korjaavien prosessien ja toimittajan varmennuksen puuttuminen heikentää entisestään erärekisterien luotettavuutta.Kun tuotantopoikkeamia ilmenee ilman vastaavaa Korjaavaa ja Ehkäisevää Toimenpidesuunnitelmaa (CAPA), eräkirjojen eheys vaarantuu [7]. Esimerkiksi, Eugia Pharma Specialities Limited, tarkastettiin 22. tammikuuta ja 2. helmikuuta 2024 välisenä aikana, ja se sai FDA 483:n, koska se ei tarkastellut poikkeamia riittävästi. Heidän tehottomat CAPA-järjestelmänsä ja puutteelliset tutkimuksensa johtivat toistuviin tuotanto-ongelmiin, mikä pakotti heidät uudistamaan kokonaan tutkimus- ja CAPA-menettelynsä [9].
Samoin, tarkastuksen aikana 26. syyskuuta ja 25. lokakuuta 2023 välillä, Stokes Healthcare Inc. osoitti huonoa poikkeamien hallintaa. Yritys ei laajentanut tutkimuksia kaikkiin vaikuttaviin eriin ja viivästytti analyysien loppuunsaattamista [9].
"Poikkeama ilman vastaavaa CAPA- tai poikkeamaraporttia? Se on vaatimustenmukaisuuden epäonnistuminen." - GXP-auditointi & Konsultointipalvelut [5]
Toimittajan varmennusongelmat lisäävät monimutkaisuutta. Empower Clinic Services LLC sai huomautuksen tarkastuksessa 18. heinäkuuta - 5. elokuuta 2022 riittämättömistä laadunvalvontamenettelyistä, mukaan lukien puutteelliset toimittajan pätevyydet ja heikot tutkimusprosessit [9]. Viljellyn lihan tuottajille, jotka ovat riippuvaisia kasvatusalustoista, solulinjoista ja muista kriittisistä syötteistä, toimittajan GMP-vaatimustenmukaisuuden varmistaminen on olennaista erärekisterien eheyden ylläpitämiseksi.
FDA:n vaatimukset erärekistereille
FDA:n säännöt erärekistereille keskittyvät 21 CFR Osa 117, joka asettaa perustason elintarviketurvallisuudelle.Kun kyseessä on viljelty liha, jossa steriliteetin ylläpitäminen soluviljelyvaiheen aikana on ratkaisevan tärkeää, dokumentoinnin on usein täytettävä tiukemmat Osa 111 tai Osa 211, standardit Osa 117 [10][14]. lisäksi. Tämä korostaa, kuinka tarkka dokumentointi on olennaista viljellyn lihan tuotannon turvallisuuden ja tehokkuuden varmistamiseksi.
Eräkohtaiset ydintandardit
Jokainen erä vaatii kaksi keskeistä asiakirjaa:
- Pääerärekisteri (MBR): Hyväksytty malli, joka kuvaa tuotantoprosessin.
- Erätuotantorekisteri (BPR): Yksityiskohtainen kirjaus siitä, mitä tuotantokierroksen aikana todella tapahtuu [12][2].
BPR:n on sisällettävä yksityiskohtia, kuten erä- tai sarjanumerot, laitetiedot, puhdistuspäivät, komponenttitunnisteet, tarkat mittaukset ja vertailut todellisten ja teoreettisten saantojen välillä [10][14].
"Erätuotantotietueen on tarkasti noudatettava asianmukaista päävalmistustietuetta, ja jokainen vaihe erän tuotannossa on suoritettava." – 21 CFR 111.255 [12]
Jokainen kriittinen vaihe on kirjattava välittömästi, ja sekä suorittajan että tarkastajan nimikirjaimet on merkittävä [10][11]. FDA vaatii ALCOA(+) -periaatteiden noudattamista, mikä tarkoittaa, että tietueiden on oltava Attribuoitavissa, Luettavissa, Ajantasaisia, Alkuperäisiä ja Tarkkoja - sekä Täydellisiä, Johdonmukaisia, Kestäviä ja Saatavilla [1].
Jos Master Manufacturing Recordista poiketaan, se on tutkittava perusteellisesti. Tämä sisältää ongelman dokumentoinnin, juurisyyn analyysin suorittamisen ja korjaavien ja ehkäisevien toimenpiteiden (CAPA) suunnitelman toteuttamisen [8] [1]. Poikkeamien alkuarvioinnit tulisi kirjata 24–48 tunnin kuluessa havaitsemisesta [8]. Elektronisia järjestelmiä käyttävissä laitoksissa 21 CFR Part 11 -vaatimusten noudattaminen on pakollista. Tämä sisältää validoidut sähköiset allekirjoitukset ja turvalliset, aikaleimatut audit trailit [8] [1].
Asiakirjojen säilytys- ja tarkistusmenettelyt
Asianmukaiset asiakirjojen säilytys- ja tarkistusprosessit ovat kriittisiä vaatimustenmukaisuuden säilyttämiseksi ja tuoteturvallisuuden varmistamiseksi.Steriilissä tuotannossa, kuten viljellyn lihan tuotannossa, jokainen yksityiskohta eräkirjanpidossa on tarkastettava huolellisesti. Laadunvalvonta (QC) -tiimi vastaa kaikkien eräkirjanpitojen tarkastamisesta, tulosten seuraamisesta ja testidatan tarkistamisesta ennen kuin erä voidaan hyväksyä jakeluun [10] [13].
"Kaikki lääketuotteiden tuotanto- ja valvontatiedot on tarkastettava ja hyväksyttävä laadunvalvontayksikön toimesta ennen erän vapauttamista tai jakelua." – 21 CFR 211.192 [2]
Valmistajat pyrkivät usein saamaan 95% erätarkastukset valmiiksi 30 päivän kuluessa tuotannosta [2] . Kuitenkin viljellyn lihan monimutkaisemmissa steriileissä prosesseissa tarkastukset vievät tyypillisesti 7–10 päivää, ja huippusuorituskykyiset laitokset saavuttavat alle 7 päivän tarkastusajat [2]. Elektroniset erärekisterijärjestelmät voivat merkittävästi nopeuttaa näitä tarkastuksia, kuten ne, jotka on integroitu viljellyn lihan tuotantojärjestelmiin, - vähentäen aikaa puoleen verrattuna paperipohjaisiin menetelmiin - kunhan ne on validoitu täyttämään osa 11 vaatimukset ja ylläpitämään tietojen eheys [1].
Mitä FDA:n hyväksymät viljellyn lihan yritykset tekivät oikein
FDA:n hyväksymät viljellyn lihan yritykset ovat asettaneet riman korkealle omaksumalla käytäntöjä, jotka vastaavat dokumentointiin liittyviin haasteisiin ja täyttävät tiukat turvallisuusstandardit.
Kun UPSIDE Foodsista tuli ensimmäinen viljellyn lihan yritys, joka läpäisi FDA:n ennakkokonsultaation marraskuussa 2022, he loivat mallin alalle.FDA antoi "ei lisäkysymyksiä" -kirjeen perusteellisen tuotantoprosessin tarkastelun jälkeen, joka sisälsi solulinjan perustamisen, solupankit, valmistuksen valvonnan sekä kaikki komponentit ja syötteet [16]. Tämä saavutus korosti yksityiskohtaisen dokumentoinnin merkitystä FDA:n tiukkojen vaatimusten täyttämisessä.
Steriliteetti- ja vaatimustenmukaisuusstandardien täyttäminen
UPSIDE Foodsin merkittävä saavutus oli heidän perusteellinen lähestymistapansa syötteiden jäljitettävyyteen. Jokainen tuotantokomponentti dokumentoitiin huolellisesti, mikä varmisti selkeän vastuun ketjun alkuperäisestä solulinjasta lopputuotteeseen [16]. Tämä läpinäkyvyyden taso mahdollisti FDA:n tarkastajien jäljittää jokaisen valmistusprosessin vaiheen, vahvistaen, että kaikki turvallisuusstandardit täytettiin johdonmukaisesti.
"FDA:n ennakkomarkkinakonsultaatio yrityksen kanssa sisälsi arvioinnin yrityksen tuotantoprosessista ja tuotantoprosessin avulla valmistetusta viljellystä solumateriaalista, mukaan lukien primaaristen ja kuolemattomien solulinjojen ja solupankkien perustaminen, valmistuksen valvonta sekä kaikki komponentit ja syötteet." – U.S. Food and Drug Administration [16]
Muut menestyneet yritykset seurasivat esimerkkiä ottamalla käyttöön yksityiskohtaisen aseptisen prosessin dokumentoinnin. Tämä sisälsi kriittisiä vaiheita, kuten pukeutumisproseduurit ja steriilit käsittelytoimet [3]. Toisin kuin aiemmat dokumentointivirheet, nämä yritykset käyttivät porrastettuja tarkistusjärjestelmiä, joihin kuului operaattoritarkastuksia, tuotannon valvontaa ja laadunvalvontayksikön tarkastuksia, jotta mahdolliset virheet havaittaisiin ennen erän vapauttamista [15]. Elektroniset erärekisterijärjestelmät olivat myös keskeisessä roolissa, varmistaen pakolliset hyväksynnät jokaisessa vaiheessa ja ylläpitäen muuttumattomia auditointijälkiä 21 CFR Part 11 -vaatimusten mukaisesti [3][2].
Nämä tiukat käytännöt ulottuivat luonnollisesti siihen, miten yritykset käsittelivät poikkeamia ja epäonnistumisia.
CAPA-prosessit eräepäonnistumisia varten
Kun erät eivät täyttäneet vaatimuksia, FDA:n hyväksymät yritykset ryhtyivät nopeisiin ja järjestelmällisiin toimiin. Heidän korjaavat ja ehkäisevät toimenpiteensä (CAPA) sisälsivät muodollisia juurisyyn analyysejä, vaikutusarviointeja ja selkeästi dokumentoituja korjaavia toimenpiteitä [3]. Kaikki poikkeamat hallittiin integroidun laadunvarmistuskehyksen sisällä, varmistaen, että kaikki ongelmat tutkittiin perusteellisesti, perusteltiin ja dokumentoitiin ennen tuotannon jatkamista [2].
Tulevaisuudessa tietojen eheys tulee olemaan FDA:n täytäntöönpanotoimien keskeinen painopiste vuosina 2024–2025 [1].
Kuinka parantaa erärekisterikäytäntöjäsi
Erärekisterikäytäntöjen vahvistaminen vaatii tarkkaa dokumentointia yleisten epäonnistumisten käsittelemiseksi, jotka usein tunnistetaan FDA:n tarkastusten aikana. Tässä on joitakin strategioita keskeisten haasteiden ratkaisemiseksi.
Suorita riippumattomia erärekisteritarkastuksia
Säännölliset kolmannen osapuolen tarkastukset voivat paljastaa ongelmia, jotka sisäiset tarkastukset saattavat jättää huomiotta. Aloita keskittymällä kriittisiin järjestelmiin, kuten laboratorion tiedonhallintajärjestelmiin (LIMS), valmistuksen ohjausjärjestelmiin (MES) ja yrityksen resurssien suunnitteluun (ERP). Priorisoi asiakirjat, joilla on suuri sääntelyvaikutus, kuten vapautustestauksen tiedot, stabiilisuustiedot ja erätuotantotiedot.
Yksi tehokas menetelmä on näytteiden noutotestaus.Valitse satunnaisesti viimeaikaisia eriä ja rekonstruoi niiden tuotanto- ja laboratoriotiedot. Tämä voi auttaa tunnistamaan puuttuvia tietoja, puutteellisia allekirjoituksia tai dokumentointiaukkoja, jotka voivat johtaa sääntelyhuomautuksiin. Ristiintarkista järjestelmän luomat audit trailit manuaalisten merkintöjen kanssa tunnistaaksesi luvattomat muutokset tai poistot.
Tarkista kaikki viime vuoden Out-of-Specification (OOS) ja Out-of-Trend (OOT) raportit. Arvioi, olivatko juurisyyanalyysit perusteellisia ja toteutettiinko Korjaavat ja Ehkäisevät Toimenpiteet (CAPA) asianmukaisesti. On syytä huomata, että dokumentointiongelmat muodostavat 21% FDA:n varoituskirjeistä, kun taas inhimillinen virhe aiheuttaa 50% erärekisteriongelmista lääketeollisuuden valmistuksessa [2] .
"Ei ole prosessin monimutkaisuus, joka laukaisee huomautuksia - vaan epäjohdonmukaisuus, puutteellisuus ja huono valvonta." – GXP-tarkastus & Konsultointipalvelut [5]
Simuloi sääntelytarkastuksia säännöllisten harjoitustarkastusten avulla. Tämä käytäntö auttaa tiimejä tunnistamaan epäjohdonmukaisuuksia ja mahdollisia tietojen eheysongelmia ennen varsinaista tarkastusta. Varmista, että kaikki asiakirjat noudattavat ALCOA+ -periaatteita: Attribuoitava, Luettava, Ajantasainen, Alkuperäinen, Tarkka, Täydellinen, Johdonmukainen, Kestävä ja Saatavilla.
Kun asiakirjojen eheys on varmistettu, keskity kaikkien tuotantopanosten laadun tarkistamiseen.
Tarkista kaikki panokset mikrobiologisen saastumisen varalta
Riippumaton steriiliys- ja tehotestaus kaikille panoksille on välttämätöntä - älä luota pelkästään toimittajan analyysitodistuksiin (CoAs). Tämä on erityisen tärkeää viljellyn lihan tuottajille, sillä saastuminen voi vaarantaa kokonaisia eriä.
Esimerkiksi helmikuussa 2013 Central Admixture Pharmacy Services sai FDA:n huomautuksia riittämättömästä mikrobikontrollista steriilien tuotteiden erien vapautuksen aikana. Yrityksen oli otettava käyttöön yksityiskohtaiset mikrobikontrollimenettelyt standardikäyttöohjeissaan (SOPs) [4].
Prosessin aikaiset mikrobikontrollipisteet voivat estää liiallisen riippuvuuden lopputuotteen testauksesta. Sisällytä nämä tarkastuspisteet erien vapautuksen SOP:ihin ja ylläpidä tiukkaa ajankohtaista dokumentointia. Kirjaa kaikki testitulokset ja valmistusvaiheet niiden tapahtuessa välttääksesi takautuvia tai viivästyneitä merkintöjä, jotka voisivat johtaa FDA:n huomautuksiin.
Pidä kattavat toimittajatiedostot, mukaan lukien CoA:t, auditointiraportit, laatusopimukset ja historia kaikista poikkeamista, jotka liittyvät saapuviin materiaaleihin.
Eräkirjauskäytäntöjen vahvistaminen edellyttää prosessien yhdenmukaistamista vakiintuneiden standardien, kuten HACCP:n ja GCCP:n, kanssa.
Yhdenmukaista asiakirjat HACCP- ja GCCP-standardien kanssa
Vaarojen analysoinnin kriittisten hallintapisteiden (HACCP) periaatteiden sisällyttäminen eräasiakirjoihin varmistaa, että kriittisiä prosessimuuttujia seurataan ja dokumentoidaan koko tuotannon ajan. Tämä sisältää prosessin aikaisen mikrobikokeilun tarkastuspisteiden perustamisen sen sijaan, että luotettaisiin pelkästään lopputestaukseen.
Viljellyn lihan tuottajille on yhtä tärkeää noudattaa hyviä soluviljelykäytäntöjä (GCCP). Eräasiakirjojen tulisi sisältää tiedot aseptisista käsittelyistä, pukeutumisprosessista ja ympäristön seurannasta, jotka liittyvät erän vapauttamiskriteereihin [3][4]. Nämä toimenpiteet auttavat ylläpitämään vaatimustenmukaisuutta ja varmistamaan tuoteturvallisuuden.
Toimialan tiedot osoittavat, että 52% dokumentointirikkomukset lisääntyvät, kun asianmukaista erävalmistusohjelmistoa ei ole käytössä [3] [4]. Esimerkki: Helmikuussa 2023 Nephron Sterile Compounding Centre sai FDA-huomautuksen, koska kriittisten prosessimuuttujien tarkistusmenettelyt puuttuivat ennen erän vapauttamista [4]. Tämä korostaa tarvetta ennakoivalle dokumentoinnille, joka on linjassa tunnustettujen standardien kanssa.
Siirtyminen sähköisiin erärekistereihin (EBR) voi merkittävästi vähentää dokumentointivirheitä - jopa 50% - reaaliaikaisen tiedonkeruun ja automatisoitujen työnkulkujen avulla [2]. Nämä järjestelmät merkitsevät puuttuvat mikrobitestitulokset tai keskeneräiset tarkastukset ennen erän etenemistä, minimoiden inhimilliset virheet.
"FDA odottaa, että tiedot ovat ALCOA(+): Attribuoitavissa, Luettavissa, Ajantasaisia, Alkuperäisiä, Tarkkoja - sekä Täydellisiä, Johdonmukaisia, Kestäviä ja Saatavilla." – Atlas Compliance [1]
Jokainen selittämätön poikkeama tai ero eräkirjanpidossa tulisi liittää viralliseen tutkimus- ja CAPA-järjestelmään. Rajoita kirjoitus- ja poistooikeuksia suojellaksesi elektronisten mikrobiologisten testitietojen eheyttä. Kilpailevat valmistajat pyrkivät tarkistamaan ja vapauttamaan 95% erät 30 päivän kuluessa tuotannon valmistumisesta [2].
Nämä toimet eivät ainoastaan vähennä huomautusten riskiä, vaan myös vastaavat FDA:n tarkastuksissa korostettuja tiukkoja dokumentaatiostandardeja.
Biopharma vs Viljelty Liha: Eroja Erätietueissa
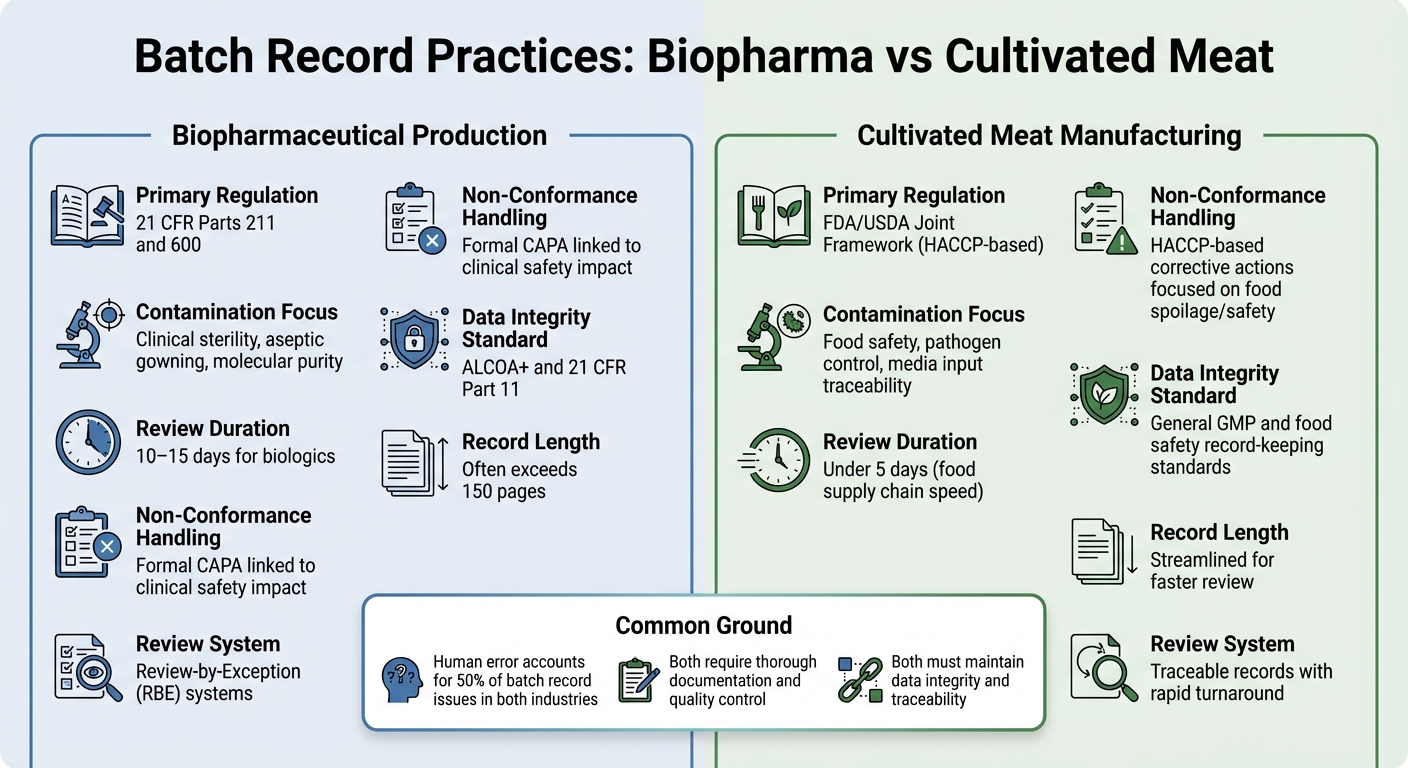
Biolääketieteellisen ja Viljellyn Lihan Erätietuevaatimusten Vertailu
Tarkasteltaessa erätietuekäytäntöjen eroja biolääketieteellisen tuotannon ja viljellyn lihan valmistuksen välillä saadaan selkeämpi kuva siitä, miten sääntelyvaatimukset muokkaavat dokumentointiprioriteetteja näillä aloilla.
Molemmat alat vaativat perusteellista dokumentointia, mutta niiden sääntelykehykset ja valvontatavoitteet eroavat merkittävästi. Biolääketieteessä erätietueet ovat tiukasti säänneltyjä 21 CFR Osat 211 ja 600, mukaan, jotka edellyttävät, että laadunvalvontayksikkö tarkistaa ja hyväksyy kaikki tuotanto- ja valvontatietueet ennen erän vapauttamista [2]. Viljellyn lihan tuottajat puolestaan noudattavat tyypillisesti HACCP ja GCCP standardeja.Nämä keskittyvät enemmän elintarviketurvallisuuteen ja patogeenien hallintaan kuin injektoitavien biologisten tuotteiden vaatimaan kliinisen tason steriiliyteen.
Biopharma-eräkirjat ovat usein laajoja, joskus yli 150 sivua, ja tarkistusprosessi voi kestää 10–15 päivää. Tämän tehostamiseksi monet biopharma-yritykset käyttävät Review-by-Exception (RBE) -järjestelmiä, jotka tiivistävät keskeiset poikkeamat kompaktiksi raportiksi. Samaan aikaan viljellyn lihan tuottajat pyrkivät jäljitettäviin asiakirjoihin, jotka voidaan tarkistaa alle viidessä päivässä, mikä heijastaa elintarvikeketjun nopeampaa tahtia [2].
Näiden asiakirjojen sisältö korostaa myös eri painopisteitä. Biopharma-tarkastukset keskittyvät usein aseptisen käsittelyn yksityiskohtiin, kuten pukeutumisprotokolliin ja ympäristön hallintaan. Sen sijaan viljellyn lihan asiakirjoissa on korostettava ravintoaineiden syöttöjä ja mikrobiologisia testejä elintarviketurvallisuuden varmistamiseksi.Viljellyn lihan osalta haasteena on monimutkaisten kasvatusalustojen seurantaan, kaikkien materiaalien mikrobiologisten testien dokumentointiin ja elintarviketurvallisuuden kriittisten rajojen täyttämiseen liittyvät vaatimukset - ilman lääketeollisuuden tiukempia steriiliysvaatimuksia.
Saastumisen ja poikkeamien trendit
| Ominaisuus | Biolääkevalmisteiden tuotanto | Viljellyn lihan valmistus |
|---|---|---|
| Pääasiallinen sääntely | 21 CFR osat 211 ja 600[2] | FDA/USDA yhteinen kehys (HACCP-pohjainen) |
| Saastumisen painopiste | Kliininen steriiliys, aseptinen pukeutuminen, molekyylipuhtaus[2] | Elintarviketurvallisuus, patogeenien hallinta, median syötteen jäljitettävyys |
| Arvioinnin kesto | 10–15 päivää biologisille tuotteille[2] | Alle 5 päivää (elintarvikeketjun nopeus) |
| Poikkeamien käsittely | Virallinen CAPA, joka liittyy kliiniseen turvallisuusvaikutukseen [2] | HACCP-pohjaiset korjaavat toimenpiteet keskittyvät elintarvikkeiden pilaantumiseen/turvallisuuteen |
| Data Integrity Standard | ALCOA+ ja 21 CFR osa 11 [1] | Yleiset GMP- ja elintarviketurvallisuuden kirjanpitostandardit |
Vaikka inhimillisten virheiden määrät ovat samankaltaisia molemmilla toimialoilla - noin 50% erärekisteriongelmista johtuu inhimillisistä virheistä [2] - panokset ovat erilaiset.Biopharmassa jopa yksi dokumentoimaton poikkeama voi aiheuttaa vakavia seurauksia potilasturvallisuudelle. Viljellyn lihan osalta kontaminaatioriskit liittyvät enemmän elintarvikeperäisiin taudinaiheuttajiin ja pilaantumiseen, mikä voi vaikuttaa koko tuotantoeriin.
Päätelmä
Eräkirjanpidot toimivat virallisena lokina jokaiselle viljellyn lihan tuotantoerälle - jos vaihetta ei ole kirjattu, sääntelyviranomaiset katsovat, että sitä ei ole suoritettu [6][3]. Tämä korostaa tarkan dokumentoinnin ja tiukan laadunvalvonnan merkitystä.
FDA:n tarkastukset korostavat, että tietojen eheyden on oltava ALCOA+ -periaatteiden mukaisia [1]. Laadunvalvontatiimien on tarkistettava ja hyväksyttävä kaikki tuotantotiedot ennen kuin erä voidaan vapauttaa [2][17], ja kaikki poikkeamat on tutkittava viipymättä dokumentoidulla juurisyyanalyysillä [2][5]. Vaikka inhimillinen virhe aiheuttaa 50% erätietojen ongelmista, kaksitasoiset tarkastukset ja rakenteelliset CAPA (Korjaavat ja ennaltaehkäisevät toimenpiteet) -prosessit voivat auttaa vähentämään näitä riskejä [2][5] .
"Ei ole prosessin monimutkaisuus, joka aiheuttaa huomautuksia - vaan epäjohdonmukaisuus, puutteellisuus ja huono valvonta." - GXP-tarkastus & Konsultointipalvelut [5]
Näiden haasteiden voittamiseksi viljellyn lihan tuottajien tulisi keskittyä riippumattomiin tarkastuksiin, elintarviketurvallisten ainesosien perusteelliseen testaukseen mikrobiologisen saastumisen varalta ja varmistaa, että dokumentaatio noudattaa HACCP- ja GCCP-standardeja. Sähköisten erärekisterijärjestelmien käyttöönotto, validoitu 21 CFR Part 11 [1], voi merkittävästi vähentää virheitä ja nopeuttaa tarkistusprosesseja.
Sääntely-ympäristö vaatii tarkkuutta, mutta se on navigoitavissa.Oppimalla biopharma-alan virheistä - kuten puuttuvista allekirjoituksista Qinhuangdao Zizhu Pharmaceutical [17], riittämättömästä kaksoisvarmennuksesta Terumo Corp [18] , ja puutteellisesta poikkeamadokumentaatiosta Torrent Pharmaceuticals [18] - viljellyn lihan yritykset voivat luoda vaatimustenmukaisia järjestelmiä alusta alkaen. Näiden oppien sisällyttäminen mahdollistaa ennakoivan vaatimustenmukaisuuden ja johdonmukaisen laadun. Turvallinen tietojen säilytys, ajantasainen poikkeamien raportointi ja realististen malliauditointien suorittaminen varmistavat, että erärekisterit ovat tarkastuskunnossa ja tuotantoerät täysin jäljitettävissä.
Lisää resursseja ja asiantuntijaohjeita viljellyn lihan valmistuksen korkeiden tuotantostandardien ylläpitämiseen saat vierailemalla
UKK
Mitä viljellyn lihan eräkirjan tulee sisältää?
Viljellyn lihan eräkirja toimii kattavana lokina koko valmistusprosessista. Sen tulee sisältää yksityiskohtaiset käsittelyohjeet, askel askeleelta suoritusmerkinnät, ja merkitä kaikki poikkeamat, jotka tapahtuvat tuotannon aikana. Lisäksi sen tulisi dokumentoida prosessin aikainen testaus ja vapautustestaus varmistaakseen, että tuote täyttää turvallisuus-, laatu- ja sääntelystandardit.
Kuinka voimme todistaa steriliteetin eräkirjojen avulla?
Steriliteetin todistaminen eräkirjojen avulla edellyttää dokumentoitujen sterilointimenetelmien, testitulosten ja väliaineen laadunvalvontaraporttien perusteellista tarkastelua varmistaakseen, että ne täyttävät sääntelyvaatimukset.On tärkeää käsitellä poikkeamat tai epäonnistuneet testit yksityiskohtaisten tutkimusten ja CAPA-toimien (Korjaavat ja ennaltaehkäisevät toimet) avulla. Tämä prosessi varmistaa, että jokainen vaihe on noudatettu ja mahdolliset ongelmat on ratkaistu asianmukaisesti steriiliysstandardien ylläpitämiseksi.
Milloin sähköisiä erärekistereitä (Osa 11) vaaditaan?
Sähköiset erärekisterit ovat välttämättömiä Osa 11:n mukaisesti, kun sähköisiä järjestelmiä käytetään dokumentoimaan, tutkimaan ja perustelemaan erärekisteripoikkeamia. Ne ovat keskeisessä roolissa varmistamassa 21 CFR Osa 211.192 , tietojen eheyden suojaamisen, tutkimusaikataulujen täyttämisen ja tehokkaan johdon valvonnan.









