

Los registros de lote son críticos para el cumplimiento y la seguridad del producto. Documentan cada paso de la producción, asegurando que se cumplan los estándares regulatorios. Para los productores de carne cultivada, mantener la esterilidad y registros detallados es innegociable. Las inspecciones de la FDA a menudo destacan problemas como datos faltantes, revisiones incompletas y acciones correctivas deficientes, lo que puede llevar a advertencias o interrupciones.
Puntos Clave:
- Registros de Lote: Dos tipos - Registro Maestro de Lote (MBR) (la "receta") y Registro de Producción de Lote (BPR) (la "ejecución").
- Problemas Comunes: Errores humanos (50% de los problemas), controles en proceso faltantes, revisiones incompletas y sistemas CAPA (Acción Correctiva y Preventiva) deficientes.
- Estándares de la FDA: La adherencia a los principios ALCOA+ (Atribuible, Legible, Contemporáneo, Original, Preciso, Completo, Consistente, Duradero, Disponible) es obligatoria.
- Soluciones: Las auditorías independientes, los registros electrónicos por lotes y la verificación rigurosa de proveedores pueden minimizar errores y mejorar el cumplimiento.
Las empresas de carne cultivada como UPSIDE Foods han establecido un punto de referencia al garantizar documentación detallada, trazabilidad de insumos y medidas correctivas rápidas. Al aprender de estas prácticas, los productores pueden evitar problemas regulatorios y mantener altos estándares de calidad.
Guía Completa de Documentación y Mantenimiento de Registros para el Cumplimiento de FDA en Ciencias de la Vida
sbb-itb-ffee270
Problemas Comunes en la Documentación de Registros por Lotes
Los informes de inspección de la FDA destacan consistentemente un problema recurrente: las desviaciones en la revisión de registros de producción se encuentran entre las principales deficiencias de GMP citadas por los reguladores [7]. Para los productores de carne cultivada, estas deficiencias van más allá de simples errores administrativos: ponen en peligro la capacidad de demostrar condiciones estériles sostenidas. Estos problemas aparecen de varias formas, como se ilustra en los ejemplos a continuación.
Revisiones Incompletas y No Conformidades
Un problema común es la falta de las Unidades de Control de Calidad para revisar a fondo los registros de lotes. En lugar de ser una parte integral del proceso de liberación, las revisiones a menudo ocurren de manera reactiva, solo después de que ya ha surgido un problema con el producto [7]. Este enfoque deja lagunas significativas en los registros de producción.
Por ejemplo, Davis City Pharmacy recibió una observación 483 de la FDA debido a registros de lotes que faltaban detalles críticos como cantidades de componentes, pasos operativos e iniciales del personal. De manera similar, CAPS fue citado por carecer de firmas requeridas y verificaciones de revisores en entradas clave [3]. Estos lapsos no son incidentes aislados; los estudios muestran que alrededor del 52% de las violaciones de documentación se agravan cuando faltan bioprocess management systems robustos [3].
"No es la complejidad del proceso lo que provoca citaciones, sino la inconsistencia, la incompletitud y la falta de supervisión adecuada." - GXP Auditing & Consulting Services [5]
Registros de Verificación en Proceso Faltantes
Otra deficiencia frecuente es la ausencia de documentación adecuada para las verificaciones en proceso. Estos registros son cruciales, especialmente en puntos de control críticos en operaciones asépticas. Por ejemplo, Nephron Sterile Compounding Center enfrentó citaciones por no documentar pasos esenciales de vestimenta y procedimientos asépticos en sus registros de lote [3]. Para los productores de carne cultivada, donde la esterilidad es primordial, tales omisiones hacen imposible confirmar el cumplimiento de medidas de control de contaminación.
Amphastar también fue señalado por no investigar o documentar variaciones inesperadas de rendimiento o discrepancias de salida [3]. Los riesgos de tales descuidos son evidentes. En un caso, se encontró que una instalación farmacéutica no identificada en 2024/2025 almacenaba registros de lotes completados en estanterías abiertas y escritorios. Los investigadores de la FDA descubrieron páginas faltantes, incluidas siete de un solo registro, y una sección completa de "Solución de Síntesis" ausente en otro [6].
Fallas en CAPA y Verificación GMP de Proveedores
Más allá de los errores de documentación, la ausencia de procesos correctivos efectivos y la verificación de proveedores socavan aún más la fiabilidad de los registros de lotes.Cuando ocurren desviaciones de producción sin un informe correspondiente de Acción Correctiva y Preventiva (CAPA), la integridad de los registros de lote se ve comprometida [7]. Por ejemplo, Eugia Pharma Specialities Limited, inspeccionada entre el 22 de enero y el 2 de febrero de 2024, recibió un FDA 483 por no revisar adecuadamente las discrepancias. Su sistema CAPA ineficaz y las investigaciones incompletas llevaron a problemas de producción repetidos, obligando a una revisión completa de sus procedimientos de investigación y CAPA [9].
De manera similar, durante una inspección del 26 de septiembre al 25 de octubre de 2023, Stokes Healthcare Inc. demostró una mala gestión de discrepancias. La empresa no extendió las investigaciones a todos los lotes afectados y retrasó la finalización de sus análisis [9].
"¿Una discrepancia sin un informe CAPA o de desviación correspondiente? Eso es un fallo de cumplimiento." - Servicios de Consultoría de Auditoría GXP & [5]
Los problemas de verificación de proveedores añaden otra capa de complejidad. Empower Clinic Services LLC fue citada durante una inspección del 18 de julio al 5 de agosto de 2022 por procedimientos de control de calidad inadecuados, incluyendo calificaciones insuficientes de proveedores y procesos de investigación deficientes [9]. Para los productores de carne cultivada, que dependen de medios de cultivo, líneas celulares y otros insumos críticos, asegurar el cumplimiento de GMP por parte de los proveedores es vital para mantener la integridad de los registros de lotes.
Requisitos de la FDA para Registros de Lotes
Las reglas de la FDA para los registros de lotes giran en torno a 21 CFR Parte 117, que establece la base para la seguridad alimentaria.Cuando se trata de carne cultivada, donde mantener la esterilidad durante la fase de cultivo celular es crucial, la documentación a menudo necesita cumplir con los estándares más estrictos de Parte 111 o Parte 211, además de la Parte 117 [10][14]. Esto subraya cómo la documentación precisa es esencial para garantizar la seguridad y efectividad de la producción de carne cultivada.
Estándares Básicos para Registros de Lotes
Cada lote requiere dos documentos clave:
- Registro Maestro de Lote (MBR): La plantilla aprobada que describe el proceso de producción.
- Registro de Producción de Lote (BPR): Un registro detallado de lo que realmente sucede durante la ejecución de la producción [12][2].
El BPR debe incluir detalles específicos como números de lote o batch, detalles del equipo, fechas de limpieza, identificadores de componentes, medidas exactas y comparaciones de rendimientos reales versus teóricos [10][14].
"El registro de producción por lotes debe seguir con precisión el registro maestro de fabricación apropiado y debe realizar cada paso en la producción del lote." – 21 CFR 111.255 [12]
Cada paso crítico debe registrarse inmediatamente, con las iniciales tanto del ejecutor como del verificador anotadas [10][11]. La FDA requiere adherirse a los principios ALCOA(+), lo que significa que los registros deben ser Atribuibles, Legibles, Contemporáneos, Originales y Exactos - así como Completos, Consistentes, Duraderos y Disponibles [1].
Si hay alguna desviación del Registro Maestro de Fabricación, debe investigarse a fondo. Esto incluye documentar el problema, realizar un análisis de la causa raíz e implementar un Plan de Acción Correctiva y Preventiva (CAPA) [8] [1]. Las evaluaciones iniciales de las desviaciones deben registrarse dentro de las 24–48 horas de su detección [8]. Para las instalaciones que utilizan sistemas electrónicos, el cumplimiento de 21 CFR Parte 11 es obligatorio. Esto incluye firmas electrónicas validadas y registros de auditoría seguros con marcas de tiempo [8] [1].
Procedimientos de Retención y Revisión de Registros
Los procesos adecuados de retención y revisión de registros son críticos para mantener el cumplimiento y garantizar la seguridad del producto.En la producción estéril, como la de carne cultivada, cada detalle en los registros de lote debe someterse a una revisión meticulosa. El equipo de Control de Calidad (QC) es responsable de revisar todos los registros de lote, monitorear los resultados y probar los datos antes de que un lote pueda ser aprobado para su distribución [10] [13].
"Todos los registros de producción y control de productos farmacéuticos deben ser revisados y aprobados por la unidad de control de calidad antes de que un lote sea liberado o distribuido." – 21 CFR 211.192 [2]
Los fabricantes a menudo apuntan a completar el 95% de las revisiones de lotes dentro de los 30 días de producción [2] . Sin embargo, para los procesos estériles más complejos involucrados en la carne cultivada, las revisiones típicamente toman 7–10 días, con instalaciones de alto rendimiento logrando tiempos inferiores a 7 días [2]. Los sistemas de registro electrónico por lotes pueden acelerar significativamente estas revisiones, como aquellos integrados en los sistemas de producción de carne cultivada, - reduciendo el tiempo a la mitad en comparación con los métodos basados en papel - siempre que estén validados para cumplir con los requisitos de la Parte 11 y mantengan la integridad de los datos[1].
Lo que hicieron bien las empresas de carne cultivada aprobadas por la FDA
Las empresas de carne cultivada aprobadas por la FDA han establecido un alto estándar al adoptar prácticas que abordan los desafíos de documentación y cumplen con rigurosos estándares de seguridad.
Cuando UPSIDE Foods se convirtió en la primera empresa de carne cultivada en pasar la consulta previa al mercado de la FDA en noviembre de 2022, establecieron un modelo para la industria.La FDA emitió una carta de "sin más preguntas" después de revisar minuciosamente su proceso de producción, que incluyó el establecimiento de líneas celulares, bancos de células, controles de fabricación y todos los componentes e insumos [16]. Este logro destacó la importancia de la documentación detallada para cumplir con los estrictos requisitos de la FDA.
Cumplimiento de Normas de Esterilidad y Conformidad
El logro destacado de UPSIDE Foods fue su enfoque exhaustivo en la trazabilidad de insumos. Cada componente de producción fue cuidadosamente documentado, asegurando una clara cadena de responsabilidad desde la línea celular inicial hasta el producto final [16]. Este nivel de transparencia permitió a los revisores de la FDA rastrear cada paso del proceso de fabricación, confirmando que se cumplieron consistentemente todos los estándares de seguridad.
"La consulta previa al mercado de la FDA con la empresa incluyó una evaluación del proceso de producción de la empresa y del material celular cultivado producido por el proceso de producción, incluyendo el establecimiento de líneas celulares primarias e inmortalizadas y bancos de células, controles de fabricación, y todos los componentes e insumos." – U.S. Administración de Alimentos y Medicamentos [16]
Otras empresas exitosas siguieron el ejemplo implementando documentación detallada de procesos asépticos. Esto incluyó pasos críticos como procedimientos de vestimenta y operaciones de manejo estéril [3]. A diferencia de los fallos de documentación anteriores, estas empresas emplearon sistemas de revisión escalonados, que involucraban verificaciones de operadores, supervisión de producción y revisiones de la unidad de calidad, para detectar posibles errores antes de la liberación del lote [15]. Los sistemas de registro electrónico por lotes también desempeñaron un papel fundamental, imponiendo aprobaciones obligatorias en cada etapa y manteniendo registros de auditoría inmutables de acuerdo con los requisitos de 21 CFR Parte 11 [3][2].
Estas prácticas rigurosas se extendieron naturalmente a cómo las empresas manejaban desviaciones y fallos.
Procesos CAPA para Fallos de Lotes
Cuando los lotes no cumplían con las especificaciones, las empresas aprobadas por la FDA tomaban medidas rápidas y sistemáticas. Sus procesos de Acción Correctiva y Preventiva (CAPA) incluían análisis formales de la causa raíz, evaluaciones de impacto y acciones correctivas claramente documentadas [3]. Cualquier desviación se gestionaba dentro de un marco integrado de aseguramiento de la calidad, asegurando que todos los problemas fueran investigados, justificados y documentados a fondo antes de continuar con la producción [2].
Mirando hacia el futuro, la integridad de los datos se convertirá en un enfoque principal de las acciones de cumplimiento de la FDA para 2024–2025 [1].
Cómo mejorar sus prácticas de registro por lotes
Fortalecer las prácticas de registro por lotes requiere una documentación precisa para abordar fallas comunes que a menudo se identifican durante las inspecciones de la FDA. Aquí hay algunas estrategias para enfrentar desafíos clave.
Realizar auditorías independientes de registros por lotes
Las auditorías regulares de terceros pueden descubrir problemas que las revisiones internas podrían pasar por alto. Comience enfocándose en sistemas críticos como los Sistemas de Gestión de Información de Laboratorio (LIMS), los Sistemas de Ejecución de Manufactura (MES) y la Planificación de Recursos Empresariales (ERP). Priorice documentos con alto impacto regulatorio, como registros de pruebas de liberación, datos de estabilidad y registros de producción por lotes.
Un método efectivo es la prueba de recuperación de muestras.Seleccione aleatoriamente lotes recientes y reconstruya su historial de producción y laboratorio. Esto puede ayudar a identificar datos faltantes, firmas incompletas o brechas de documentación que podrían llevar a citaciones regulatorias. Verifique las pistas de auditoría generadas por el sistema con las entradas manuales para identificar cambios o eliminaciones no autorizadas.
Revise todos los informes de Fuera de Especificación (OOS) y Fuera de Tendencia (OOT) del último año. Evalúe si los análisis de causa raíz fueron exhaustivos y si las Acciones Correctivas y Preventivas (CAPAs) se implementaron adecuadamente. Cabe destacar que los problemas de documentación representan el 21% de las cartas de advertencia de la FDA, mientras que el error humano contribuye al 50% de los problemas de registros de lotes en la fabricación farmacéutica [2].
"No es la complejidad del proceso lo que desencadena citaciones, sino la inconsistencia, la incompletitud y la mala supervisión." – GXP Auditing & Servicios de Consultoría [5]
Simule inspecciones regulatorias a través de revisiones simuladas periódicas. Esta práctica ayuda a los equipos a reconocer inconsistencias y posibles problemas de integridad de datos antes de una auditoría real. Asegúrese de que todos los registros sigan los principios ALCOA+: Atribuible, Legible, Contemporáneo, Original, Preciso, Completo, Consistente, Duradero y Disponible.
Una vez que la integridad de la documentación sea sólida, concéntrese en verificar la calidad de todos los insumos de producción.
Pruebe Todos los Insumos para Contaminación Microbiológica
Las pruebas independientes de esterilidad y potencia para todos los insumos son esenciales - no confíe únicamente en los Certificados de Análisis (CoAs) del proveedor. Esto es particularmente crucial para los productores de carne cultivada, ya que la contaminación puede poner en peligro lotes enteros.
Por ejemplo, en febrero de 2013, Central Admixture Pharmacy Services enfrentó citaciones de la FDA debido a un control microbiano inadecuado durante la liberación de lotes de productos estériles. La empresa tuvo que introducir procedimientos detallados de control microbiano en sus Procedimientos Operativos Estándar (SOPs) [4].
Los puntos de control microbiano en proceso pueden prevenir la dependencia excesiva de las pruebas del producto final. Incorpore estos puntos de control en los SOPs de liberación de lotes y mantenga una documentación estricta y contemporánea. Registre todos los resultados de las pruebas y los pasos de fabricación a medida que ocurren para evitar registros retroactivos o entradas retrasadas, lo que podría resultar en citaciones de la FDA.
Mantenga archivos completos de proveedores, incluidos CoAs, informes de auditoría, acuerdos de calidad y un historial de cualquier desviación relacionada con los materiales entrantes.
El fortalecimiento de las prácticas de registro de lotes implica además alinear los procesos con estándares establecidos como HACCP y GCCP.
Alinear Registros con Estándares HACCP y GCCP
Incorporar los principios de Análisis de Peligros y Puntos Críticos de Control (HACCP) en los registros de lotes garantiza que las variables críticas del proceso sean monitoreadas y documentadas durante toda la producción. Esto incluye establecer puntos de control de pruebas microbianas en proceso en lugar de depender únicamente de pruebas en la etapa final.
Para los productores de carne cultivada, la adherencia a los estándares de Buenas Prácticas de Cultivo Celular (GCCP) es igualmente vital. Los registros de lotes deben incluir detalles de manipulaciones asépticas, procedimientos de vestimenta y monitoreo ambiental vinculados a los criterios de liberación de lotes [3][4]. Estos pasos ayudan a mantener el cumplimiento y garantizar la seguridad del producto.
Los datos de la industria muestran que el 52% de las violaciones de documentación aumentan cuando no se cuenta con un software adecuado de fabricación por lotes [3][4]. Un caso concreto: en febrero de 2023, el Centro de Compuestos Estériles Nephron recibió una observación de la FDA debido a la ausencia de procedimientos de control para verificar las variables críticas del proceso antes de la liberación del lote [4]. Esto resalta la necesidad de una documentación proactiva alineada con estándares reconocidos.
La transición a Registros Electrónicos de Lotes (EBR) puede reducir significativamente los errores de documentación - hasta en un 50% - mediante la recopilación de datos en tiempo real y flujos de trabajo automatizados [2]. Estos sistemas señalan resultados de pruebas microbiológicas faltantes o revisiones incompletas antes de que un lote avance, minimizando el error humano.
"La FDA espera que los registros sean ALCOA(+): Atribuibles, Legibles, Contemporáneos, Originales, Exactos - además de Completos, Consistentes, Duraderos y Disponibles." – Atlas Compliance [1]
Cada discrepancia o desviación inexplicada en los registros de lotes debe estar vinculada a un sistema formal de investigación y CAPA. Limite los permisos de escritura y eliminación para proteger la integridad de los datos de pruebas microbiológicas electrónicas. Los fabricantes competitivos buscan revisar y liberar el 95% de los lotes dentro de los 30 días posteriores a la finalización de la producción [2].
Estas acciones no solo reducen el riesgo de citaciones, sino que también se alinean con los rigurosos estándares de documentación destacados en las recientes inspecciones de la FDA.
Biopharma vs Carne Cultivada: Diferencias en los Registros de Lote
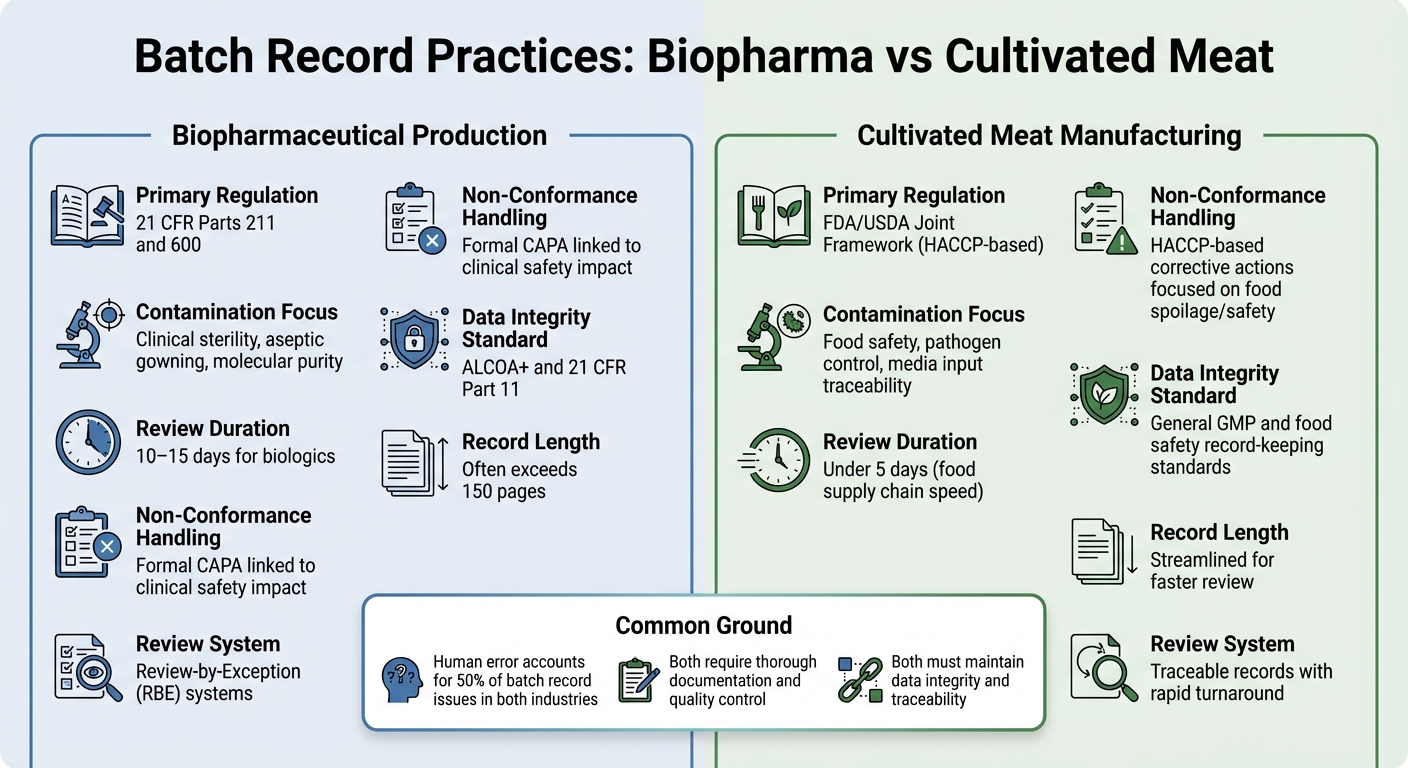
Comparación de Requisitos de Registros de Lote entre Biopharmaceutical y Carne Cultivada
Examinar las diferencias en las prácticas de registros de lote entre la producción biofarmacéutica y la fabricación de carne cultivada ofrece una imagen más clara de cómo las demandas regulatorias moldean las prioridades de documentación en estas industrias.
Ambos sectores requieren documentación exhaustiva, pero sus marcos regulatorios y objetivos de control difieren significativamente. En biofarmacéutica, los registros de lote están estrictamente regulados bajo 21 CFR Partes 211 y 600, que requieren que la unidad de Control de Calidad revise y apruebe todos los registros de producción y control antes de que un lote pueda ser liberado [2]. Los productores de carne cultivada, por otro lado, típicamente siguen los estándares HACCP y GCCP.Estos se centran más en la seguridad alimentaria y el control de patógenos, en lugar de la esterilidad de grado clínico exigida para los biológicos inyectables.
Los registros por lotes de biopharma suelen ser extensos, a veces superando las 150 páginas, y el proceso de revisión puede llevar de 10 a 15 días. Para agilizar esto, muchas empresas de biopharma utilizan sistemas de Revisión por Excepción (RBE), que resumen las desviaciones clave en un informe compacto. Mientras tanto, los productores de carne cultivada buscan registros rastreables que puedan revisarse en menos de cinco días, reflejando el ritmo más rápido de la cadena de suministro de alimentos [2].
El contenido de estos registros también resalta las diferentes prioridades. Las inspecciones de biopharma a menudo se centran en los detalles del procesamiento aséptico, como los procedimientos de vestimenta y los controles ambientales. En contraste, los registros de carne cultivada deben enfatizar los insumos de medios y las pruebas microbiológicas para garantizar la seguridad alimentaria.Para la carne cultivada, el desafío radica en rastrear entradas de medios complejos, documentar pruebas microbiológicas para todos los materiales y cumplir con los límites críticos de seguridad alimentaria, sin adherirse a los requisitos de esterilidad más estrictos de los productos farmacéuticos.
Tendencias de Contaminación y No Conformidad
| Característica | Producción Biofarmacéutica | Fabricación de Carne Cultivada |
|---|---|---|
| Regulación Primaria | 21 CFR Partes 211 y 600 [2] | Marco Conjunto FDA/USDA (basado en HACCP) |
| Enfoque de Contaminación | Esterilidad clínica, vestimenta aséptica, pureza molecular [2] | Seguridad alimentaria, control de patógenos, trazabilidad de insumos de medios |
| Duración de la Revisión | 10–15 días para biológicos [2] | Menos de 5 días (velocidad de la cadena de suministro de alimentos) |
| Manejo de No Conformidades | CAPA formal vinculada al impacto en la seguridad clínica [2] | Acciones correctivas basadas en HACCP enfocadas en el deterioro/seguridad alimentaria |
| Estándar de Integridad de Datos | ALCOA+ y 21 CFR Parte 11 [1] | Estándares generales de GMP y registro de seguridad alimentaria |
Aunque las tasas de error humano son similares en ambas industrias - alrededor del 50% de los problemas de registros de lotes surgen de errores humanos [2] - las implicaciones son diferentes.En biopharma, incluso una sola desviación no documentada podría tener serias implicaciones para la seguridad del paciente. Para la carne cultivada, los riesgos de contaminación están más relacionados con patógenos transmitidos por alimentos y el deterioro, lo que puede afectar lotes de producción enteros.
Conclusión
Los registros de lotes sirven como el registro oficial para cada lote de producción de carne cultivada: si un paso no está registrado, los reguladores consideran que no se realizó [6][3]. Esto resalta la importancia de la documentación precisa y el control de calidad estricto.
Las inspecciones de la FDA enfatizan que la integridad de los datos debe alinearse con los principios ALCOA+ [1]. Los equipos de Control de Calidad deben revisar y aprobar todos los registros de producción antes de que un lote pueda ser liberado [2][17], y cualquier desviación debe ser investigada de inmediato con un análisis de causa raíz documentado [2][5]. Aunque el error humano representa el 50% de los problemas en los registros de lotes, las revisiones de doble nivel y los procesos estructurados de CAPA (Acción Correctiva y Preventiva) pueden ayudar a reducir estos riesgos [2][5].
"No es la complejidad del proceso lo que provoca citaciones, sino la inconsistencia, la incompletitud y la mala supervisión." - Servicios de Consultoría de Auditoría GXP & [5]
Para superar estos desafíos, los productores de carne cultivada deben centrarse en auditorías independientes, pruebas rigurosas de ingredientes seguros para alimentos para la contaminación microbiológica, y asegurar que la documentación cumpla con los estándares HACCP y GCCP. Implementar sistemas de registro electrónico de lotes, validados bajo 21 CFR Parte 11 [1], puede minimizar significativamente los errores y acelerar los procesos de revisión.
El entorno regulatorio exige precisión, pero es navegable.Al aprender de los errores de la biofarmacéutica, como las firmas faltantes en Qinhuangdao Zizhu Pharmaceutical [17], verificación dual insuficiente en Terumo Corp [18] , y documentación de desviaciones inadecuada en Torrent Pharmaceuticals [18], las empresas de carne cultivada pueden establecer sistemas conformes desde el principio. Incorporar estas lecciones permite un cumplimiento proactivo y una calidad constante. La retención segura de registros, la notificación oportuna de desviaciones y la realización de auditorías simuladas realistas garantizarán que los registros de lotes estén listos para inspección y que las producciones sean completamente rastreables.
Para más recursos y orientación experta sobre cómo mantener altos estándares de producción en la fabricación de carne cultivada, visite
Preguntas Frecuentes
¿Qué debe incluir un registro de lote para carne cultivada?
Un registro de lote para carne cultivada sirve como un registro completo de todo el proceso de fabricación. Debe incluir instrucciones de procesamiento detalladas, registros de ejecución paso a paso, y anotar cualquier desviación que ocurra durante la producción. Además, debe documentar pruebas en proceso y pruebas de liberación para confirmar que el producto cumple con los estándares de seguridad, calidad y regulación.
¿Cómo podemos demostrar la esterilidad usando registros de lote?
Demostrar la esterilidad a través de registros de lote implica examinar minuciosamente los procedimientos de esterilización documentados, los resultados de las pruebas y los informes de control de calidad de los medios para asegurar que cumplan con los requisitos regulatorios.Es crucial abordar cualquier desviación o prueba fallida a través de investigaciones detalladas y CAPAs (Acciones Correctivas y Preventivas). Este proceso asegura que cada paso se haya seguido y que cualquier problema se haya resuelto adecuadamente para mantener los estándares de esterilidad.
¿Cuándo se requieren registros electrónicos de lotes (Parte 11)?
Los registros electrónicos de lotes son esenciales bajo la Parte 11 cuando se utilizan sistemas electrónicos para documentar, investigar y justificar las desviaciones de los registros de lotes. Desempeñan un papel crítico en asegurar el cumplimiento con 21 CFR Parte 211.192 , protegiendo la integridad de los datos, cumpliendo con los plazos de investigación y asegurando una supervisión efectiva de la gestión.









