

Validering av tillväxtmedium är ett obligatoriskt steg för företag inom odlat kött som söker godkännande på den brittiska marknaden. Denna process säkerställer produkternas säkerhet, kvalitet och efterlevnad av strikta regleringsramar som Storbritanniens Novel Food Regulations (EU 2015/2283). Här är vad du behöver veta:
- Viktiga krav: Tillväxtmedium måste uppfylla standarder för toxikologi, kontaminationskontroll, näringskvalitet och allergenicitet.
- Brittiska regleringar: Food Standards Agency (FSA) kräver efterlevnad av HACCP-principer och klassificering under Produkter av animaliskt ursprung (POAO).
- Globala standarder: Medan Storbritannien och EU delar liknande ramar, följer USA CGMP-regler under FD&C Act.
- Valideringsprocess: Inkluderar noggrann testning av sammansättning, renhet, funktionalitet och leverantörskompatibilitet, tillsammans med robust dokumentation.
- Stödinitiativ: Storbritanniens regulatoriska sandlåda på £1,6 miljoner, lanserad 2025, hjälper företag att uppfylla dessa standarder.
Korrekt validering säkerställer säkerhet, bygger förtroende och överensstämmer med lagkrav. Artikeln går djupare in i steg-för-steg-processen, inklusive testmetoder, leverantörskvalifikationer och tips för regulatoriska inlämningar.
Regulatoriska standarder för tillväxtmedia
Standarder och riktlinjer
Tillväxtmedia, en kritisk komponent i odlat köttproduktion, måste uppfylla strikta internationella regulatoriska standarder. Dessa standarder varierar mellan regioner, var och en med specifika krav på sammansättning, säkerhet och renhet.
I Storbritannien regleras odlingsmedia under Novel Food Regulations (assimilerad förordning (EU) 2015/2283). Innan de godkänns för marknaden krävs en grundlig säkerhetsbedömning [1]. Food Standards Agency (FSA) klassificerar cellodlade produkter som produkter av animaliskt ursprung (POAO) enligt förordning (EG) 853/2004. Denna klassificering kräver att producenter implementerar livsmedelssäkerhetssystem baserade på principerna för faroanalys och kritiska styrpunkter (HACCP) [3]. FSA håller också på att utveckla detaljerad teknisk vägledning om sammansättningen av odlingsmedia, med ytterligare uppdateringar förväntade [1]. Dessa ramar utgör grunden för mer specifika regleringskrav.
I USA skiljer sig tillvägagångssättet.Tillväxtmediekomponenter måste uppfylla kraven för Current Good Manufacturing Practice (CGMP) som beskrivs i avsnitt 501(a)(4)(B) i Federal Food, Drug, and Cosmetic Act (FD&C Act) [4]. FDA kategoriserar mediekomponenter som "förnödenheter och reagenser", vilka regleras av 21 CFR delarna 210 och 211. Dessa komponenter måste genomgå kvalitetsverifiering för att förhindra kontaminering [4]. Intressant nog klassificeras syntetiska komponenter av odlat köttmedia - såsom aminosyror, vitaminer och salter - ofta som Klass I medicintekniska produkter under 21 CFR 864.2220, vilket undantar dem från krav på förhandsanmälan [6][7].
I Europeiska unionen är det regulatoriska ramverket nära anpassat till Storbritanniens, eftersom det också följer förordning (EU) 2015/2283.The Europeiska myndigheten för livsmedelssäkerhet (EFSA) övervakar godkännandeprocessen [1]. Enligt ICH Q6B-riktlinjer behandlas tillväxtmediekomponenter, inklusive antibiotika, inducerare och andra beståndsdelar, som processrelaterade föroreningar. Dessa föroreningar måste kontrolleras och reduceras till acceptabla nivåer [5]. Där det är möjligt bör hjälpämnen och reagenser uppfylla farmakopéstandarder [5].
| Jurisdiktion | Primär reglering | Klassificering | Säkerhetssystem | Medieövervakning |
|---|---|---|---|---|
| Storbritannien (GB) | Assimilerad förordning (EU) 2015/2283 [1] | Produkt av animaliskt ursprung (POAO) [3] | HACCP (Förordning 852/2004) [3] | FSA/FSS Sandbox-vägledning [1] |
| Europeiska unionen / NI | Förordning (EU) 2015/2283 [1] | Produkt av animaliskt ursprung (POAO) [3] | HACCP (Förordning 852/2004) [3] | EFSA-auktorisationsprocess [1] |
| United States | FD&C Act Section 501(a)(4)(B) [4] | Ny djurläkemedel / mat [4] | CGMP (21 CFR 210/211) [4] | FDA CVM / USDA-FSIS [4] |
Regulatoriska krav för odlat kött
Producenter av odlat kött måste säkerställa att varje sats av tillväxtmedium uppfyller strikta säkerhets- och kvalitetsstandarder.Tillväxtmedievalidering är en viktig aspekt av den bredare regleringsramen för dessa produkter. Enligt HACCP-principerna (Förordning (EG) 852/2004) identifieras tillväxtmedium som en primär insats och en potentiell källa till kontaminering - kemisk, mikrobiell eller annan [3]. FSA framhäver denna oro:
"De främsta farorna vid produktion av cellodlade produkter rör cellinjeidentitet (och konsistens), faror som introduceras under produktionsprocessen (mikrobiologisk kontaminering, tillväxtmedium och restkomponenter i slutprodukten) och allergener." [3]
Om det sker förändringar i tillväxtmediets formulering krävs en omedelbar HACCP-översyn [3].I Storbritannien måste validering ske innan implementering för att säkerställa noggrannheten i flödesscheman och effektiviteten av kontrollåtgärder [3].
I USA kräver FDA att alla reagenser och mediekomponenter uppfyller strikta kvalitetsstandarder för att undvika att skadliga ämnen introduceras [4]. Leverantörer och kontraktslaboratorier måste följa CGMP-föreskrifter, och alla leverantörer som inte följer dessa bör tas bort för att förhindra att produkter klassificeras som "förfalskade" [4]. FDA betonar vikten av detta:
"Alla nya veterinärläkemedel, inklusive ACTP:er, måste tillverkas i enlighet med CGMP för att säkerställa att sådana läkemedel uppfyller kraven i Federal Food, Drug, and Cosmetic Act (FD&C Act) vad gäller säkerhet." [4]
För närvarande samarbetar flera företag som deltar i Storbritanniens regulatoriska sandlåda - såsom BlueNalu, Gourmey, Hoxton Farms, Mosa Meat, Roslin Technologies, Vital Meat, och Vow - med FSA för att förfina dessa tekniska standarder [1]. Enligt brittiska regler kan företag begära upp till fem års dataskydd för konfidentiell information som lämnas in under godkännandeprocessen [1].
Steg för validering av tillväxtmedium
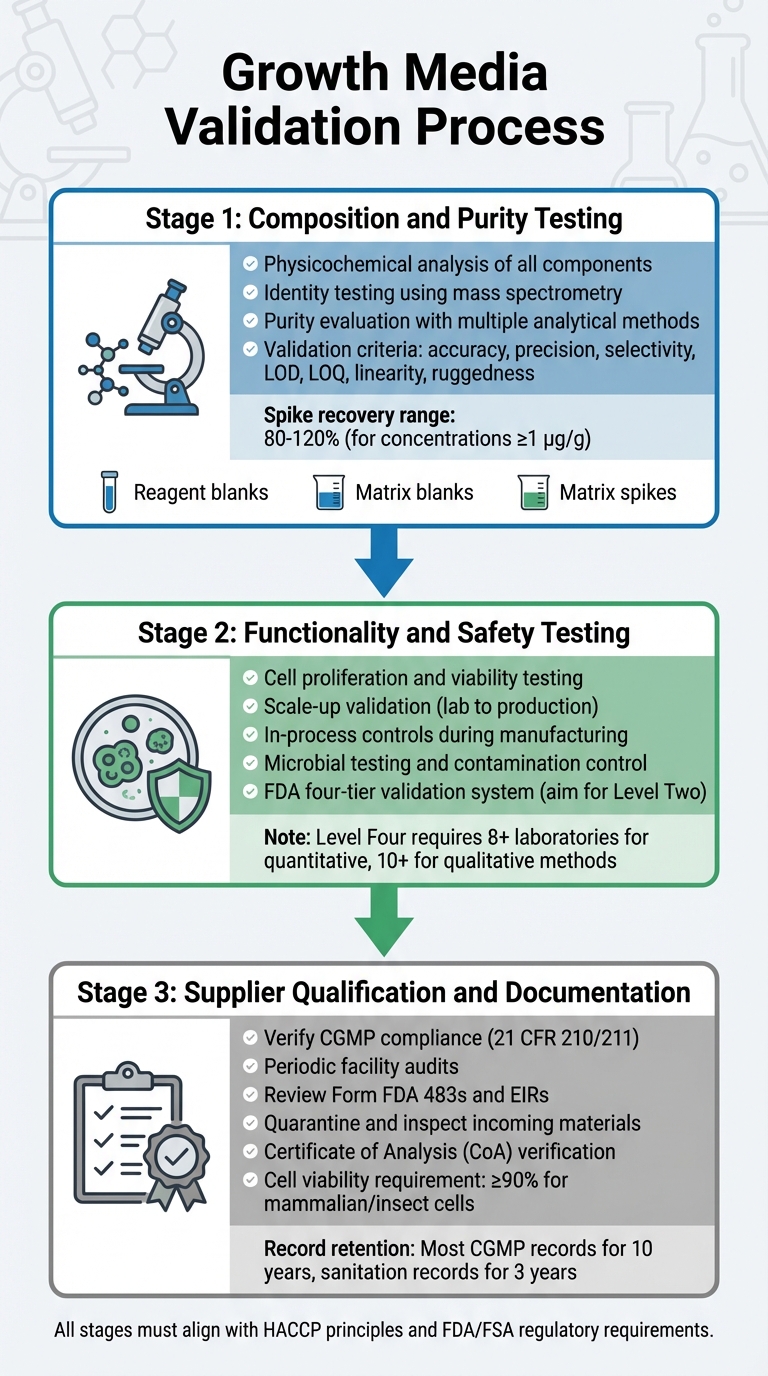
Valideringsprocess för tillväxtmedium för odlat kött för regulatoriskt godkännande
Validering av tillväxtmedium innebär en detaljerad process som undersöker dess sammansättning, funktionalitet, säkerhet och leverantörskompatibilitet. Varje steg bygger på det föregående, vilket säkerställer en robust valideringsprocess som överensstämmer med regulatoriska krav. Detta inkluderar testning av sammansättning, funktionalitet och leverantörskompatibilitet.
Testning av sammansättning och renhet
Det första steget fokuserar på en grundlig fysikalisk-kemisk analys av varje komponent. Detta innebär att identifiera den exakta sammansättningen, de fysiska egenskaperna och molekylstrukturen hos ingredienser som aminosyror, vitaminer och oorganiska salter [5].För att bekräfta molekylstrukturer använder identitetstestning mycket specifika metoder, inklusive fysikalisk-kemiska, biologiska och immunokemiska tekniker. Verktyg som masspektrometri används för att verifiera molekylära identiteter genom deras fragmenteringsmönster [8].
Renhetsutvärdering kräver flera analytiska metoder för att separera önskade komponenter från föroreningar. Dessa tester måste ta itu med både processrelaterade och produktrelaterade föroreningar [5]. Analytiska tekniker bör uppfylla strikta valideringskriterier, inklusive noggrannhet, precision, selektivitet, detektionsgränser (LOD), kvantifieringsgränser (LOQ), linearitet och robusthet [8]. Valideringsprotokoll bör också inkludera:
- Reagensblanka för att säkerställa att reagenser är fria från analyter.
- Matrix blanks för att bekräfta att provmiljön inte stör.
- Matrix spikes för att uppskatta återvinning och noggrannhet.
För kvantitativa metoder vid koncentrationer av 1 µg/g (ppm), är acceptabla spikåtervinningar vanligtvis mellan 80% och 120% [8].
För att upprätthålla konsekvens bör tillverkare etablera interna primära referensmaterial hämtade från produktionsrepresentativa partier. Dessa fungerar som spårbara standarder för kalibrering av arbetsreferensmaterial [5]. När renhetstestningen är klar måste mediet visa sin förmåga att stödja effektiv celltillväxt och uppfylla säkerhetsstandarder.
Funktionalitet och säkerhetstestning
Efter att ha bekräftat sammansättningen måste mediet bevisa sin effektivitet i att stödja produktion av odlat kött. Detta inkluderar att visa att celler kan föröka sig, bibehålla livskraft och skala från laboratorieförhållanden till produktionsvolymer. FDA kräver processkontroller under tillverkningen, från tidiga stadier som cellpassage och skörd, för att säkerställa produktens konsistens och säkerhet [4].
Säkerhetsvalidering involverar rigorös mikrobiell testning och kontaminationskontroll, enligt FDA:s förhandsutvärderingar [9].
FDA använder ett fyrstegssystem för kemisk metodvalidering, från nivå ett (nödsituation eller begränsad användning) till nivå fyra (fullständiga samarbetsstudier som uppfyller AOAC/ISO-standarder) [8].För rutinmässig regulatorisk testning, sikta på Nivå Två enkel-laboratorie validering, som inkluderar en omfattande prestandautvärdering [8]. Fullständiga samarbetsstudier för kvantitativa metoder kräver deltagande från minst åtta laboratorier, medan kvalitativa metoder behöver tio [8]. När mediets prestanda är validerad, är det viktigt att säkerställa att alla råmaterial kommer från godkända leverantörer.
Leverantörskvalificering och Dokumentation
Tillverkare måste arbeta med verifierade, CGMP-kompatibla leverantörer. Leverantörer bör uppfylla de standarder som anges i 21 CFR 210/211 [4]. Verifiering innebär periodiska revisioner av leverantörsanläggningar för att bedöma efterlevnad av kvalitetsprogram, procedurer och övergripande CGMP-efterlevnad [4].
Innan du ingår avtal, granska en leverantörs efterlevnadshistorik, inklusive Form FDA 483s och Establishment Inspection Reports (EIRs) [4]. FDA betonar denna skyldighet:
"Innan du ingår något kontrakt, avtal eller annan överenskommelse med en annan anläggning för att utföra något tillverkningssteg åt dig, bör du verifiera att anläggningen följer tillämpliga regler för CGMP." [4]
Alla inkommande material måste sättas i karantän och inspekteras innan de släpps, för att säkerställa att de uppfyller huvudspecifikationerna [10]. Leverantörer är skyldiga att tillhandahålla ett analyscertifikat (CoA) eller spårbara, CGMP/GLP-kompatibla testresultat [10].För stabila cellinjer måste dokumentationen inkludera en spårbar kloningshistorik [10]. Däggdjurs- eller insektsceller kräver vanligtvis minst 90% livskraft för att accepteras i CGMP-projekt [10]. Register bör behållas enligt regulatoriska riktlinjer [4].
Kontrakt måste tydligt ange CGMP-ansvar och kräva att leverantörer meddelar tillverkare om eventuella föreslagna ändringar av testkit eller metoder [4]. Om testning är outsourcad, säkerställ att kontraktslaboratorier använder validerade analytiska metoder och är FDA-registrerade [4].
Förbereda dokument för regulatorisk inlämning
När ditt tillväxtmedium har validerats är nästa steg att sammanställa en dossier som visar att alla säkerhets- och kvalitetsstandarder som krävs av FDA och USDA-FSIS uppfylls. Denna dossier fungerar som en kritisk länk mellan validering och regulatorisk efterlevnad, vilket ger myndigheterna en tydlig bild av ditt mediums säkerhet och produktionsprocesser.
Nödvändiga element i en inlämningsdossier
Din dossier bör innehålla en detaljerad uppdelning av mediets sammansättning, med en lista över alla aminosyror, vitaminer, oorganiska salter och tillväxtfaktorer. FDA:s riktlinjer betonar att granskningsprocessen inte bara utvärderar själva mediet utan hela produktionsarbetsflödet. Detta inkluderar etablering av cellinjer och banker, implementering av tillverkningskontroller och verifiering av alla komponenter och insatser [11].
Dessutom måste dossiern innehålla en grundlig säkerhets- och toxikologisk bedömning som bevisar livsmedelssäkerheten för det odlade materialet och alla dess insatser. Inkludera tillverkningskontrollregister, processvalideringsdata och dokumentation av kvalitetsprogram för att visa att din produktion är konsekvent och fri från föroreningar.
Du bör också tillhandahålla verifieringsregister för leveranser och reagenser, som visar validering för alla material som används i mediet, inklusive de som förbereds internt. För produkter som regleras av USDA-FSIS, inkludera HACCP-planer och saneringsprotokoll. FDA rekommenderar att de flesta CGMP-register behålls i minst 10 år, medan register över anläggningsrengöring och sanering bör behållas i minst 3 år [4]. Detta överensstämmer med leverantörskvalificeringsinsatser, vilket säkerställer att alla insatser uppfyller CGMP och regulatoriska krav.
Dokumentation av anläggningens efterlevnad
Innan produktion, bearbetning eller lagring av odlat kött för mänsklig konsumtion måste anläggningar registrera sig hos FDA [12]. Din dokumentation bör inkludera en omfattande livsmedelssäkerhetsplan som behandlar faroanalys (biologiska, kemiska och fysiska), förebyggande åtgärder (såsom sanitet, allergenhantering och åtgärder i leveranskedjan) och tillsynsförfaranden [12].
Mediefyllnadssimuleringar är också ett viktigt krav. Dessa involverar 14-dagars inkubation och tillväxtfrämjande tester för att bekräfta aseptiska metoder.Som FDA förklarar:
"Mediefyllningen bör utvärdera den aseptiska monteringen och driften av den kritiska (sterila) utrustningen, kvalificera operatörerna och bedöma deras teknik, samt visa att miljökontrollerna är tillräckliga" [2].
Säkerställ att dina register inkluderar leverantörskvalificeringsdata, såsom tester utförda på de första tre satserna av medium från en leverantör för att bekräfta att de matchar analyscertifikatet. Andra viktiga register inkluderar loggar för miljökontroll, utrustningskalibreringsscheman och temperaturövervakningsdata. För USDA-reglerade processer, förbered HACCP-planer, skriftliga sanitära standardrutiner (SSOPs) och återkallelseprocedurer [12][13].
sbb-itb-ffee270
Använda Cellbase för regleringskompatibel upphandling av tillväxtmedia
Verifierade leverantörer för odlat kött
När du har validerat din tillväxtmediaformulering är nästa steg att skaffa komponenter som uppfyller regulatoriska standarder. Detta är inte lika enkelt som att beställa från generiska leverantörer. För cellodlade produkter gäller strikta hygienregler, och varje komponent i tillväxtmediet måste komma med specifik dokumentation för regulatoriskt godkännande [3]. Det är där
Inköpsfunktioner
Plattformen erbjuder också transparent prissättning och en direktmeddelandefunktion, vilket gör det möjligt för team att snabbt begära offerter, analyscertifikat och andra regulatoriska dokument.Genom att konsolidera dessa kritiska upphandlingsfunktioner i ett system anpassat för produktion av odlat kött, förenklar
Slutsats
Att validera tillväxtmedia för regulatoriskt godkännande är inte bara en formalitet - det är ett lagkrav för att introducera odlade köttprodukter på den brittiska marknaden. Detta innebär noggranna tester för sammansättning och renhet, implementering av en stark HACCP-plan och att hålla detaljerad dokumentation genom hela processen.
"Mat får inte släppas ut på marknaden om den är osäker. Detta innebär att den varken är skadlig för hälsan eller olämplig för mänsklig konsumtion." - Food Standards Agency [3]
The UK Food Standards Agency's £1.6 million Regulatory Sandbox betonar sitt engagemang för att samarbeta med branschaktörer för att etablera tydliga tekniska riktlinjer för tillväxtmediasammansättning [1]. Företag som prioriterar korrekt validering nu kommer att vara i en starkare position när dessa riktlinjer är fullt definierade.
Att uppfylla efterlevnadsstandarder handlar inte bara om att kryssa i regulatoriska rutor - det handlar om att vinna konsumenternas förtroende och säkerställa produktsäkerhet. Strikt kvalitetstestning är kärnan i både regulatoriskt godkännande och att få marknadsacceptans. För att effektivisera godkännandeprocessen, fokusera på att bygga starka valideringsprotokoll, upprätthålla korrekta register och samarbeta med pålitliga leverantörer. Dessa steg kommer inte bara att förenkla godkännandet utan också bana väg för större konsumentförtroende.
Vanliga frågor
Vilka är de viktigaste stegen för att validera tillväxtmedia för regulatoriskt godkännande?
Att validera tillväxtmedia för regulatoriskt godkännande handlar om att bevisa att formuleringen är säker, pålitlig och lämplig för att producera odlat kött. Så här ser processen vanligtvis ut:
- Riskbedömning: Börja med att definiera cellinjen du kommer att använda, produktens mål och dess kritiska kvalitetsattribut (som pH eller näringssammansättning). Identifiera eventuella potentiella faror, såsom mikrobiell kontaminering, och lägg upp åtgärder för att kontrollera dessa risker.
- Testning och specifikationer: Sätt tydliga acceptanskriterier för faktorer som sterilitet, renhet och styrka. Använd etablerade testmetoder för att säkerställa att dessa standarder konsekvent uppfylls.
- Valideringsstudier: Genomför noggrann processvalidering, inklusive kvalificering av utrustning och testning av flera batcher, för att bekräfta att resultaten är reproducerbara och konsekventa.
- Stabilitetstestning: Kontrollera hur mediet håller över tid genom att bedöma dess kvalitet under hela dess avsedda hållbarhetstid under korrekta lagringsförhållanden (vanligtvis 2–8 °C).
- Dokumentation: Samla allt i en omfattande valideringsdossier. Detta bör inkludera alla testresultat och analyser för att uppfylla regulatoriska krav.
Genom att noggrant adressera varje steg, kommer du att samla in de bevis som behövs för att visa att mediet uppfyller de säkerhets- och kvalitetsstandarder som krävs för produktion av odlat kött.
Vilka är de viktigaste skillnaderna mellan brittiska och amerikanska regler för tillväxtmedia som används i odlat kött?
I Storbritannien regleras tillväxtmedia för odlat kött under Novel Foods Regulation (EU Regulation 2015/2283), som har behållits i brittisk lag. Alla tillväxtmedia som används i produkter som inte vanligtvis konsumerades före den 15 maj 1997 måste genomgå en formell bedömning av nya livsmedel av Food Standards Agency (FSA). Denna process kräver inlämning av detaljerad dokumentation, inklusive information om mediets sammansättning, ursprung och renhet. Dessutom krävs en HACCP-baserad riskbedömning för att visa hur föroreningar kontrolleras under cellodlingsprocessen.
Sedan december 2025 har FSA implementerat en Cell-Cultivated Products sandbox. Detta initiativ erbjuder vägledning och stödjer snabbare datainsamling för ansökningar om nya livsmedel.För att få slutgiltigt godkännande måste företag lämna in en omfattande dossier som behandlar mediesäkerhet, konsekvens och tillverkningsvalidering. Endast efter detta godkännande kan produkten säljas i Storbritannien.
I kontrast har USA inte ett specifikt ramverk för nya livsmedel anpassat för tillväxtmedier, vilket gör direkta regulatoriska jämförelser utmanande. För företag baserade i Storbritannien kan det förenkla godkännandeprocessen att skaffa mediekomponenter som redan uppfyller dessa strikta standarder.Hur stödjer Storbritanniens regulatoriska sandlåda validering av tillväxtmedia?
Storbritanniens regulatoriska sandlåda för odlade produkter erbjuder en välorganiserad miljö där företag kan testa och förfina sina tillväxtmedieformuleringar. Under överinseende av Food Standards Agency (FSA) och Food Standards Scotland (FSS) körs detta program i sexmånadersfaser. Under denna tid kan företag genomföra säkerhetstester, utföra riskbedömningar och granska dokumentation samtidigt som de får värdefull feedback från tillsynsmyndigheter.
Detta praktiska tillvägagångssätt möjliggör praktiska försök och steg-för-steg-förbättringar, vilket påskyndar insamlingen av säkerhetsdata och hjälper företag att anpassa sig till regulatoriska krav. För de som arbetar med odlat kött kan sourcing av förgodkända tillväxtmedier genom









