

La validación de salas limpias garantiza que los entornos de producción cumplan con estrictos estándares de contaminación, vitales para la producción de carne cultivada. Este es un paso crítico al escalar los procesos de carne cultivada. La validación adecuada previene riesgos de contaminación, protege la calidad del producto y cumple con regulaciones como ISO 14644 y GMP. El proceso involucra cuatro fases clave:
- Calificación de Diseño (DQ): Confirma que el diseño de la sala limpia cumple con las necesidades operativas y regulatorias.
- Calificación de Instalación (IQ): Verifica que los componentes estén instalados correctamente y cumplan con las especificaciones.
- Calificación Operacional (OQ): Prueba los sistemas en un estado inactivo para asegurar que funcionen como se espera.
- Calificación de Desempeño (PQ): Evalúa el desempeño de la sala limpia durante la producción real.
Los protocolos de prueba, incluidos los conteos de partículas, las verificaciones de integridad del filtro HEPA y las mediciones de flujo de aire, son críticos para mantener el cumplimiento. El monitoreo continuo y la revalidación periódica ayudan a mantener el rendimiento de la sala limpia a lo largo del tiempo. Seguir estos pasos asegura que los riesgos de contaminación se minimicen, protegiendo tanto la consistencia del producto como la aprobación regulatoria.
Validación de Salas Limpias desde URS hasta PQ
sbb-itb-ffee270
Las 4 Fases de la Validación de Salas Limpias
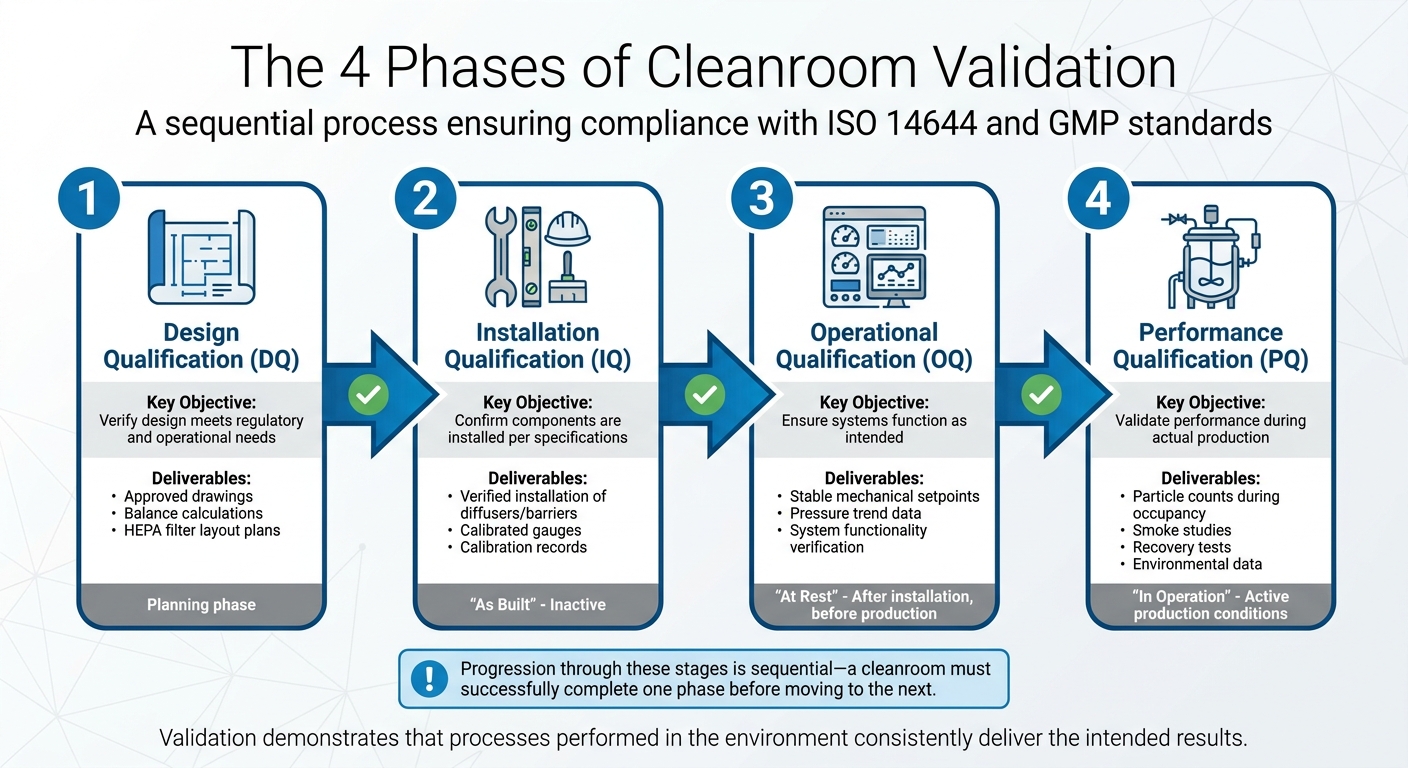
4 Fases de la Validación de Salas Limpias para la Producción de Carne Cultivada
La validación de salas limpias es un proceso paso a paso con cuatro fases distintas, cada una construyendo sobre la anterior. La progresión a través de estas etapas es secuencial: una sala limpia debe completar con éxito una fase antes de pasar a la siguiente.Como afirma acertadamente Allied Cleanrooms:
"La validación es lo que separa una sala limpia que parece lista de una que realmente lo está" [8].
Mientras que la calificación asegura que la sala limpia y sus sistemas están instalados y funcionan según lo diseñado, la validación va un paso más allá. Demuestra que los procesos realizados en el entorno entregan consistentemente los resultados previstos [7]. Las cuatro etapas - Calificación de Diseño (DQ), Calificación de Instalación (IQ), Calificación Operacional (OQ) y Calificación de Desempeño (PQ) - están diseñadas para preparar las instalaciones para procesos de producción validados. Estas etapas también sientan las bases para protocolos de prueba rigurosos.
| Fase de Validación | Objetivos Clave | Entregables/Pruebas Típicas |
|---|---|---|
| Calificación de Diseño (DQ) | Verificar que el diseño cumpla con las necesidades regulatorias y operativas. | Dibujos aprobados, cálculos de balance, planes de disposición de filtros HEPA. |
| Calificación de Instalación (IQ) | Confirmar que los componentes están instalados según las especificaciones. | Instalación verificada de difusores/barreras, calibración de medidores. |
| Calificación Operacional (OQ) | Asegurar que los sistemas funcionen como se espera. | Puntos de ajuste mecánicos estables, datos de tendencia de presión. |
| Calificación de Desempeño (PQ) | Validar el desempeño durante la producción/ocupación. | Conteo de partículas, estudios de humo, pruebas de recuperación, datos de monitoreo ambiental. |
Calificación de Diseño (DQ)
La fase de Calificación de Diseño (DQ) asegura que el diseño de la sala limpia se alinea con los requisitos específicos de la producción de carne cultivada. Esto implica verificar que los documentos de diseño, como los cálculos de balance y los diseños de filtros HEPA, reflejen las necesidades operativas reales. Cada elemento de diseño debe cumplir con criterios de aceptación estrictos, a menudo basados en los estándares ISO 14644 o en requisitos definidos por el usuario [7].
Calificación de Instalación (IQ)
La Calificación de Instalación (IQ) se centra en verificar la condición "tal como se construyó" de la sala limpia en su estado inactivo. Esta fase confirma que los difusores, retornos y barreras coinciden con las especificaciones de diseño. También verifica que los monitores de presión y los medidores estén correctamente calibrados y completamente operativos.Documentación detallada, incluidos los registros de calibración y las ubicaciones de prueba mapeadas, es crítica para esta etapa [7][8].
Calificación Operacional (OQ)
La Calificación Operacional (OQ) prueba la sala limpia en su estado "en reposo" - después de la instalación pero antes de que comience la producción. Esta fase asegura que los sistemas funcionen como se espera documentando puntos de ajuste mecánicos estables y tendencias de presión consistentes. Si ocurren cambios significativos, como reubicar equipos o modificar el flujo de aire, se requiere una nueva prueba específica para mantener el equilibrio [7][8]. Una vez que se confirma que los sistemas operan correctamente, la instalación está lista para la validación de rendimiento bajo condiciones activas.
Calificación de Rendimiento (PQ)
La etapa final, Calificación de Rendimiento (PQ), valida el rendimiento de la sala limpia durante condiciones de producción reales.Esta fase evalúa si la instalación cumple con los objetivos de rendimiento mientras se utiliza para la producción de carne cultivada. Las evaluaciones clave incluyen conteos de partículas durante la ocupación, visualización del flujo de aire (como estudios de humo) alrededor de áreas críticas y pruebas de recuperación para medir qué tan rápido la sala vuelve a la limpieza requerida después de una perturbación. Antes de comenzar PQ, asegúrese de que los puntos de ajuste mecánicos sean estables, gestionados a través de bioprocess control software, se identifiquen ubicaciones críticas de muestreo y los registros de limpieza confirmen condiciones validadas [7].
Para instalaciones de carne cultivada, se recomienda encarecidamente utilizar agencias de validación independientes de terceros. Esta verificación imparcial tiene más peso con los reguladores y auditores. Allied Cleanrooms enfatiza:
"Los reguladores y auditores dan más peso a los resultados que provienen de una parte externa sin interés en el resultado" [8].
Este enfoque independiente es especialmente crucial para las instalaciones que buscan una USDA concesión de inspección, que requiere la finalización exitosa de la consulta previa a la comercialización de la FDA [5] [6].
Protocolos de Pruebas Requeridos para la Validación de Salas Limpias
Una vez que se completan la Calificación de Diseño (DQ), la Calificación de Instalación (IQ), la Calificación Operacional (OQ) y la Calificación de Desempeño (PQ), el siguiente paso es una serie exhaustiva de pruebas para verificar el rendimiento de la sala limpia. Estas pruebas aseguran que la sala limpia cumpla con su clasificación ISO y sea apta para la producción de carne cultivada. A continuación se presenta una visión general de los protocolos de prueba clave.
Pruebas de Conteo de Partículas en el Aire
Esta prueba mide el número de partículas en el aire para confirmar que la sala limpia se adhiere a su clasificación ISO.Por ejemplo, una sala limpia ISO 5 no debe exceder 3,520 partículas de 0.5 µm o más grandes por metro cúbico. Las pruebas implican el uso de contadores de partículas calibrados en puntos de muestreo designados bajo condiciones tanto "en reposo" como "en operación". Según ISO 14644-2, las pruebas de concentración de partículas deben realizarse cada seis meses para las clasificaciones ISO 5 y más estrictas, y anualmente para ISO 6 y superiores [8].
Pruebas de Integridad de Filtros HEPA
Estas pruebas aseguran que los filtros de aire particulado de alta eficiencia (HEPA) funcionen correctamente, sin fugas ni defectos. Mientras que las pruebas de conteo de partículas evalúan la limpieza general de la sala, las pruebas de integridad se centran en los propios filtros. Cualquier cambio significativo, como reemplazos de filtros o modificaciones en la sala, requiere una nueva prueba inmediata.Muchas instalaciones optan por agencias de terceros para llevar a cabo estas pruebas, ya que la verificación independiente a menudo es muy valorada por los reguladores [8].
Mediciones de Velocidad y Volumen de Flujo de Aire
El flujo de aire adecuado es crítico para mantener la limpieza. El flujo de aire en salas limpias unidireccionales debería típicamente estar dentro de 0.45 m/s ±20% (entre 0.36 y 0.54 m/s). Las mediciones generalmente se toman a la altura de trabajo - donde ocurren operaciones sensibles, como la inoculación de biorreactores, dentro de sistemas de biorreactores escalables - o de 150 a 300 mm desde la cara del filtro. La norma ISO 14644-3:2005 establece que el número de puntos de muestreo debe ser igual a la raíz cuadrada de 10 veces el área de la sala (en metros cuadrados), con un mínimo de cuatro lecturas y al menos un punto por filtro.Los estudios de humo o la visualización del mapeo del flujo de aire pueden verificar aún más el flujo de aire unidireccional y detectar áreas de aire estancado, conocidas como "regiones de estela" [9] .
Verificaciones de Diferencial de Presión
Mantener diferenciales de presión adecuados entre las zonas de salas limpias es esencial para prevenir la contaminación. Las zonas más limpias deben mantener una presión positiva en relación con las áreas adyacentes menos limpias. Se utilizan manómetros y sensores calibrados para documentar y asegurar diferenciales de presión estables.
Verificación de Temperatura y Humedad
Los niveles de temperatura y humedad de la sala limpia deben controlarse cuidadosamente para apoyar la producción de carne cultivada. Estas condiciones influyen en la calidad del producto, así como en el rendimiento de los filtros HEPA y otros sistemas. La monitorización continua ayuda a garantizar que estos parámetros permanezcan dentro de los puntos de ajuste requeridos durante los ciclos de producción.
Monitoreo Continuo y Revalidación
La validación no se detiene una vez que los sistemas están en su lugar. El monitoreo continuo y la revalidación periódica son esenciales para contrarrestar los efectos del desgaste de los filtros, la degradación del sistema HVAC y los cambios en el proceso. Después de lograr el cumplimiento inicial a través de DQ, IQ, OQ y PQ, mantener el rendimiento durante la producción activa requiere supervisión continua.
Programas de Monitoreo Ambiental
Un programa robusto de monitoreo ambiental realiza un seguimiento de los conteos de partículas en el aire, la contaminación microbiana, la temperatura, la humedad y las diferencias de presión según un cronograma definido. Para las zonas de Grado A, el monitoreo debe ser continuo, mientras que las zonas de Grado B requieren verificaciones cada 15–30 minutos. Las zonas de Grado C y D pueden ser monitoreadas por hora o por turno, basándose en evaluaciones de riesgo [3][4].
El monitoreo microbiano combina el muestreo activo de aire con placas de sedimentación. Según las directrices de GMP del Reino Unido, las placas de sedimentación deben probarse al menos semanalmente, mientras que los conteos de partículas no viables deben realizarse diariamente. La frecuencia de monitoreo debe aumentar después de las actividades de mantenimiento [3][4]. Todos los datos deben registrarse en tiempo real, con límites de alerta definidos. Por ejemplo, una zona de Grado A podría establecer un límite de acción de 1 UFC/m³ para partículas viables [1][2]. Analizar las tendencias en estos datos puede ayudar a identificar problemas potenciales temprano.
Herramientas avanzadas como contadores de partículas láser remotos, muestreadores de aire activos y registradores de datos con alertas en tiempo real aseguran un monitoreo continuo.Las redes de sensores inalámbricos proporcionan supervisión 24/7 a través de paneles de control, reduciendo la dependencia de verificaciones manuales [2][10]. Para mantener la precisión, los sensores deben someterse a mantenimiento preventivo cada seis meses.
Programación de Revalidación
La revalidación asegura que el rendimiento de la sala limpia se mantenga dentro de las especificaciones requeridas, incluso a medida que el equipo envejece, los procesos evolucionan o cambian los requisitos regulatorios. Los desencadenantes para la revalidación incluyen cambios importantes, como la instalación de nuevos biorreactores, la actualización de sistemas HVAC o la modificación de diseños de instalaciones. Para las instalaciones de carne cultivada, los cambios de proceso - como modificaciones en la formulación de medios - también deben tenerse en cuenta para gestionar los riesgos de contaminación [1] [3].
Los parámetros críticos deben ser revalidados anualmente, con verificaciones semestrales y revalidación inmediata tras cambios significativos. Según las directrices GMP de MHRA, las salas limpias de alto riesgo para carne cultivada deben revalidar su Calificación de Desempeño (PQ) cada 12 meses, cubriendo todos los elementos IQ, OQ y PQ. Después de las actualizaciones de HVAC, las pruebas deben realizarse dentro de los 30 días [4] [10]. Los horarios de mantenimiento preventivo también deben alinearse con las auditorías GMP [2][3].
Para las necesidades de validación continua,
Estándares de Cumplimiento para Salas Limpias de Carne Cultivada
Después de abordar los protocolos de validación y prueba, el obstáculo final para la producción de carne cultivada es cumplir con los estándares de cumplimiento para asegurar la aprobación regulatoria. Las salas limpias utilizadas en este proceso deben adherirse a ISO 14644 para límites de partículas y métodos de prueba, junto con las directrices de Buenas Prácticas de Manufactura (GMP) para el control de contaminación y validación. Siguiendo estos marcos, los fabricantes pueden asegurar que sus instalaciones cumplan con las estrictas demandas regulatorias. Desglosaremos el papel de cada estándar en el cumplimiento de salas limpias.
ISO 14644 Normas para la Clasificación de Salas Limpias
ISO 14644 describe las clasificaciones de salas limpias basadas en la concentración de partículas en el aire. Mide partículas de tamaño ≥ 0.5 μm por metro cúbico, con clases que van desde ISO 1 (la más limpia) hasta ISO 9. Para la producción de carne cultivada, las clasificaciones más relevantes son ISO 5 a ISO 8, que se alinean con los Grados GMP A a D. Estas normas se centran en condiciones "en reposo", cuando la sala limpia está completamente configurada pero desocupada.
Mientras que ISO 14644 establece la base para clasificar salas limpias, no cubre la validación durante operaciones activas ni requiere monitoreo microbiano. Aquí es donde entran en juego las directrices GMP, añadiendo una capa adicional de cumplimiento para las instalaciones de carne cultivada.
Requisitos de GMP para Carne Cultivada
A diferencia de las normas ISO, GMP exige validación tanto para los estados "en reposo" (desocupado) como "en operación" (ocupado). Por ejemplo, una sala limpia de Grado B permite hasta 3,520 partículas ≥ 0.5 μm/m³ cuando está en reposo, pero esto aumenta a 352,000 partículas durante la operación [12] .
GMP emplea una Estrategia de Control de Contaminación (CCS), guiada por la Gestión de Riesgos de Calidad (QRM), para identificar y minimizar los riesgos de contaminación. Las directrices también especifican requisitos estructurales y de superficie para prevenir la acumulación de partículas y permitir una limpieza efectiva. Las superficies deben ser lisas, impermeables y duraderas, mientras que se desaconsejan las puertas correderas debido a las dificultades de limpieza. Además, se prohíben los fregaderos y desagües en las áreas de Grado A y B para evitar reservorios microbianos.
Dado que los humanos son responsables del 75-80% de las partículas detectadas durante las inspecciones de salas limpias [11], GMP impone estrictos protocolos de vestimenta y limita el acceso del personal durante las fases críticas de Calificación de Desempeño (PQ).
Para productos que requieren manejo estéril, la validación GMP incluye simulaciones de procesos asépticos (llenados de medios) para confirmar que el proceso de producción puede prevenir la contaminación microbiana. El monitoreo ambiental es otro aspecto crítico, cubriendo tanto partículas no viables como microorganismos viables. Las zonas de Grado A requieren monitoreo continuo, mientras que las áreas de grado inferior se someten a controles frecuentes para mantener el cumplimiento.
Uso de Cellbase para Recursos de Validación de Salas Limpias
La adquisición de equipos de validación de salas limpias para instalaciones de carne cultivada puede ser un proceso complicado, principalmente debido a las herramientas de monitoreo especializadas necesarias para cumplir con los estándares ISO 14644 y GMP. Las plataformas generales de suministro de laboratorios a menudo no tienen estos artículos especializados, dejando a los equipos de adquisiciones a juntar soluciones de redes de proveedores fragmentadas. Entra
Acceso a Equipos y Materiales Verificados
Tome, por ejemplo, una startup que redujo exitosamente su cronograma de validación al abastecerse a través de
Procurement Simplificado para Necesidades de Validación
Además de ofrecer equipos verificados,
Los gerentes de adquisiciones han informado un reabastecimiento más rápido de herramientas de monitoreo esenciales, incluidos contadores de partículas en tiempo real y registradores de datos, que son clave para mantener programas efectivos de monitoreo ambiental y programar la revalidación bajo las directrices de GMP [18] . Además,
Conclusión
La validación de salas limpias en la producción de carne cultivada es un proceso meticuloso diseñado para asegurar que las instalaciones cumplan con los límites de partículas ISO 14644 y los estándares GMP antes de que puedan comenzar las operaciones del biorreactor. Los datos hablan por sí mismos: las salas limpias validadas consistentemente logran una tasa de garantía de esterilidad del 99.99%, con instalaciones que cumplen con ISO 14644 reportando tasas de contaminación por debajo del 1%.En contraste, los entornos no validados enfrentan tasas de contaminación de hasta el 15% - una diferencia notable que resalta la importancia de una validación adecuada[13] [14].
Pero el trabajo no se detiene después de la validación inicial. Mantener el rendimiento de la sala limpia es igual de importante. Según expertos del Instituto de Tecnología de Salas Limpias, la validación inadecuada representa el 40% de las no conformidades GMP en biofarmacéutica. Para la carne cultivada, esto representa un riesgo serio, ya que incluso un solo evento de contaminación podría poner en peligro lotes de producción por valor de decenas de miles de libras, destacando la necesidad de una capa de adquisición confiable para asegurar insumos de alta calidad[13][14].
Preguntas Frecuentes
¿Cuál es la diferencia entre calificación y validación en una sala limpia?
La calificación y la validación desempeñan roles diferentes pero igualmente importantes en el mantenimiento del cumplimiento de la sala limpia.
Calificación se trata de asegurar que la sala limpia y sus sistemas estén correctamente instalados y funcionen como se espera. Este proceso involucra varias etapas, incluyendo Calificación de Diseño (DQ), Calificación de Instalación (IQ), y Calificación Operacional (OQ). Cada paso confirma que la sala limpia cumple con sus especificaciones de diseño y opera de manera efectiva.
Validación, por otro lado, se enfoca en la capacidad de la sala limpia para proporcionar consistentemente el ambiente requerido durante la producción real. Se trata de asegurar la fiabilidad a largo plazo, la seguridad y el cumplimiento de las normas regulatorias.
¿Cómo elijo la clase ISO/Grado GMP adecuado para áreas de carne cultivada?
Al elegir la clase ISO o el grado GMP adecuado para la producción de carne cultivada, todo se reduce a la etapa específica de producción y los riesgos de contaminación asociados.
- Clase ISO 5: Mejor para las etapas tempranas de cultivo donde mantener la esterilidad es crucial.
- Clase ISO 6: Ideal para operaciones de biorreactores, equilibrando la limpieza con la practicidad.
- Clase ISO 8: Adecuada para procesos de cosecha y transferencia, donde los riesgos de contaminación son menores.
Mantener estándares de limpieza más altos es esencial en áreas donde la esterilidad no puede ser comprometida. Además, los controles ambientales adecuados son imprescindibles para cumplir con los requisitos regulatorios.
¿Qué cambios requieren una revalidación inmediata de la sala limpia?
Cuando se realizan cambios importantes, como alteraciones en el diseño de la sala limpia, la incorporación de nuevos equipos o actualizaciones en los controles ambientales que podrían influir en la esterilidad o el cumplimiento, se hace necesaria una revalidación inmediata. Tales cambios pueden afectar condiciones críticas, por lo que la revalidación asegura que todo continúe cumpliendo con los requisitos regulatorios.










