

Jeśli najpierw edytujesz, a potem sprawdzasz, możesz naprawić zmianę poza celem w klonie. Utrzymałbym prosty przepływ pracy: wybierz metodę edycji o najniższym ryzyku, skróć czas ekspozycji edytora, a następnie przetestuj zarówno przewidywane miejsca poza celem, jak i stabilność klonu przed wydaniem.
Dla inżynierów bioprocesów, naukowców zajmujących się hodowlą komórek i zespołów R&D zajmujących się mięsem hodowlanym, główny punkt jest prosty. Systemy CRISPR mogą nadal ciąć w miejscach o bliskim dopasowaniu, często tolerując 3–6 niedopasowań, a te błędy mogą przenosić się do rozszerzonych klonów jednokomórkowych. Artykuł dzieli kontrolę ryzyka na trzy fazy: przed edycją, w trakcie edycji, i po edycji.
Oto pełna lista kontrolna w prostych słowach:
-
Wybierz narzędzie edycyjne o najniższym ryzyku do zadania
- Użyj edycji bazowej lub edycji pierwotnej, gdy mogą one dostarczyć edycję bez przerwania podwójnej nici
- Użyj modulacji opartej na dCas9, jeśli potrzebujesz tylko regulacji genów
- Jeśli potrzebujesz nukleazy, zacznij od wysokiej wierności wariantu Cas9
-
Zabezpiecz materiał wyjściowy
- Potwierdź tożsamość linii komórkowej
- Sprawdź mykoplazmę
- Zapisz numer pasażu
- Sekwencjonuj rzeczywiste miejsce docelowe w linii roboczej, a nie tylko genom referencyjny
-
Przesiej przewodniki przed pracą mokrą
- Użyj narzędzi opartych na wyrównaniu i opartych na ocenach do analizy efektów ubocznych razem
- Preferuj przewodnik z czystszym profilem efektów ubocznych niż taki z wyższą aktywnością na celu
- Zwróć uwagę na długość przewodnika, 40–60% zawartości GC, i sekwencje homopolimerowe
-
Ogranicz ekspozycję wewnątrz komórki
- Użyj RNP lub mRNA zamiast systemów plazmidowych lub wirusowych, jeśli to możliwe
- Użyj minimalnej skutecznej dawki
- Unikaj przedłużania trwałości edytora tylko po to, aby wymusić wyniki transfekcji
-
Dodaj dodatkowe kontrole dla przypadków o wyższym ryzyku
- Rozważ sparowane nukleazy
- Użyj indukowalnych, split-Cas9, lub systemów kontrolowanych światłem, gdy czas ma znaczenie
- Dodaj białka anty-CRISPR jako krok wyłączający, gdy jest to potrzebne
-
Zweryfikuj poprawnie po edycji
- Najpierw potwierdź edycję na docelowym miejscu
- Sprawdź każde przewidywane miejsce poza celem za pomocą ukierunkowanego NGS ampliconów
- Przejdź do GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, lub WGS, gdy ryzyko projektu jest wyższe
- Dla edytorów bazowych lub edytorów pierwotnych, dodaj kontrole na poziomie RNA , gdzie to istotne
-
Nie wypuszczaj pojedynczego klonu tylko na podstawie sekwencji
- Porównaj 2–3 niezależne klony
- Użyj nieedytowanej kontroli rodzicielskiej
- Usuń klony z niestabilnością lub dryfem fenotypowym
- Wypuść tylko wtedy, gdy stan edycji, ekran poza celem i zapisy są kompletne
Krótki sposób myślenia o tym: projektowanie w celu uniknięcia niezamierzonych cięć, dostarczanie w celu ograniczenia czasu w komórce, a następnie walidacja na głębokości, którą uzasadnia ryzyko projektu. To jest wątek przewijający się przez cały utwór.
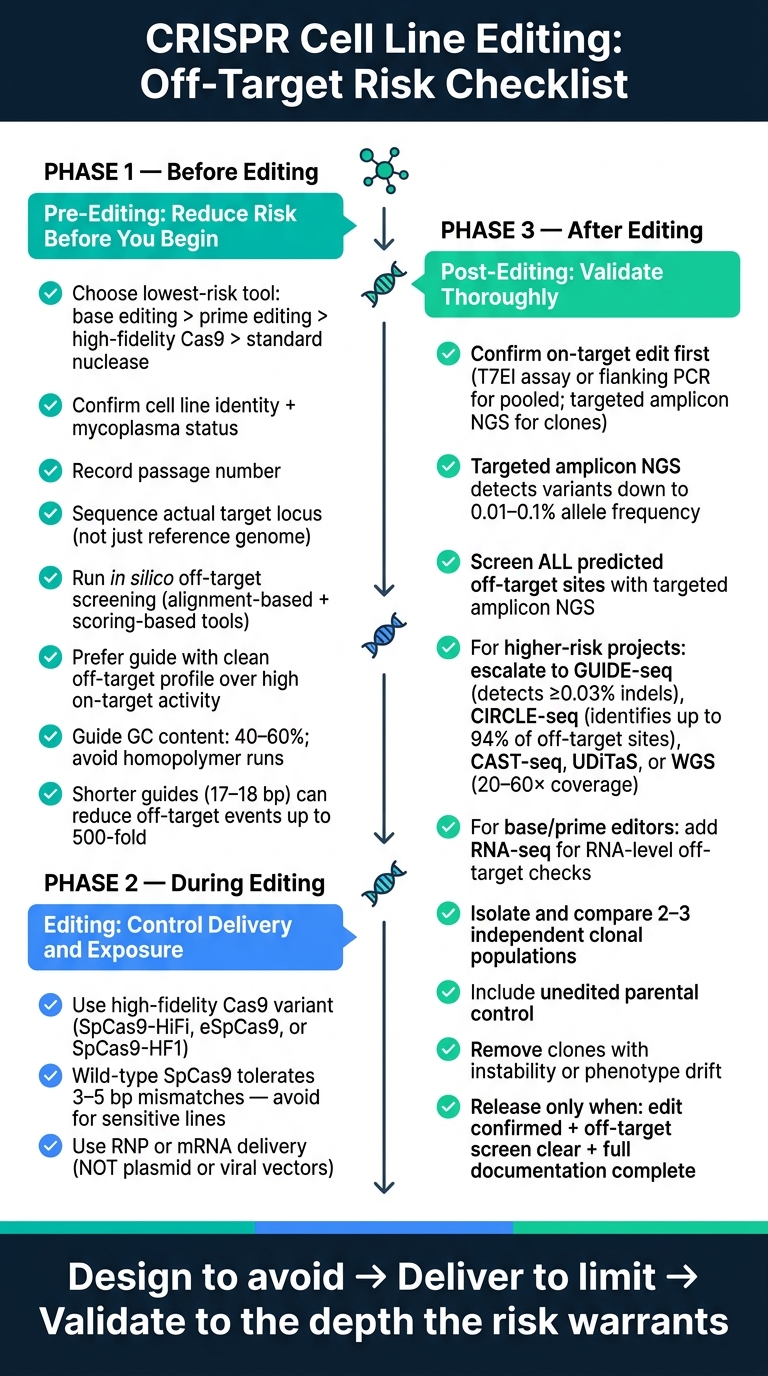
Kontrola ryzyka poza celem CRISPR: 3-etapowa lista kontrolna dla edycji linii komórkowych
Lista kontrolna przed edycją: zmniejsz ryzyko przed rozpoczęciem edycji
Zdefiniuj cel edycji i wybierz metodę edycji o najniższym ryzyku
Zanim zamówisz pojedynczy odczynnik, dokładnie określ, co ma na celu edycja. Nokaut, wstawienie, zmiana pojedynczego nukleotydu i modulacja transkrypcyjna nie niosą tego samego ryzyka poza celem. Nie wymagają również tego samego narzędzia.
Ogólna kolejność ryzyka jest prosta. Nukleazy tworzące DSB, takie jak Cas9 i Cas12 , znajdują się na najwyższym poziomie ryzyka, ponieważ mogą powodować duże delecje, translokacje i odpowiedzi na uszkodzenia DNA [1] [7]. Edytory bazowe i edytory pierwotne używają nickaz, więc unikają DSB i zmniejszają ryzyko zmienności strukturalnej [1][5]. Dla modulacji transkrypcyjnej, edytory epigenetyczne takie jak dCas9 połączone z modyfikatorami transkrypcyjnymi pozostawiają sekwencję DNA niezmienioną [1].
Zasada praktyczna jest prosta: używaj najmniej genotoksycznej metody, która nadal może dostarczyć potrzebną edycję. Dla zmian pojedynczych nukleotydów, CBEs lub ABEs są lepszym wyborem niż HDR, które nadal może wprowadzać indels [3] [1]. Dla substytucji oraz małych insercji lub delecji, edytowanie pierwotne często wykazuje niższą aktywność poza celem niż standardowy CRISPR-Cas9 [1]. Jeśli musisz użyć nukleazy, wybierz wariant o wysokiej wierności, taki jak SpCas9-HiFi, eSpCas9, lub SpCas9-HF1 [1] [6].
Gdy podejście do edycji jest ustalone, zabezpiecz linię komórkową i dokładną sekwencję docelową.
Potwierdź tożsamość linii komórkowej, historię i sekwencję locus docelowego
Jeśli linia komórkowa jest błędnie zidentyfikowana lub zanieczyszczona krzyżowo, reszta przepływu pracy zaczyna się chwiać. Nawet dobrze zaprojektowany RNA przewodnikowy nie uratuje złego materiału wyjściowego. Sprawdź tożsamość linii komórkowej przed rozpoczęciem jakiejkolwiek edycji. Jednocześnie potwierdź status mykoplazmy i zanotuj aktualny numer pasażu, ponieważ komórki o wysokim pasażu mogą zmieniać stabilność genomu i wydajność edycji [1][6].
Równie ważne jest, aby nie polegać wyłącznie na genomie referencyjnym.Sekwencjonuj dokładny docelowy locus w linii komórkowej. Ten krok pomaga zidentyfikować SNP lub indels, które mogą blokować wiązanie przewodnika lub tworzyć nowe miejsca poza celem [1] [6].
Następnie przejdź do projektowania przewodnika.
Przeprowadź in silico screening poza celem przed wyborem odczynników
Gdy docelowy locus zostanie potwierdzony, przeprowadź in silico screening kandydatów RNA przewodników przed przystąpieniem do pracy laboratoryjnej. Użyj zarówno narzędzi opartych na dopasowaniu, takich jak Cas-OFFinder lub FlashFry, oraz narzędzi opartych na ocenach, takich jak ocena CFD lub DeepCRISPR . Pierwsza grupa pomaga znaleźć miejsca genomowe z homologami sekwencji. Druga pomaga uszeregować te miejsca według przewidywanego prawdopodobieństwa cięcia [1][5].
Podczas wybierania przewodników, czystszy profil poza celem powinien przewyższać surową efektywność na celu. Przewodnik z 70% efektywnością na celu i bez przewidywanych poza celów jest bezpieczniejszym punktem wyjścia niż taki z 90% efektywnością i kilkoma miejscami wysokiego ryzyka [6]. W niektórych przypadkach skrócenie długości przewodnika z 20 bp do 17-18 bp może zmniejszyć zdarzenia poza celem nawet 500-krotnie bez większej utraty dokładności na celu [5]. Dąż do zawartości GC między 40% a 60% i unikaj serii czterech lub więcej identycznych zasad [6][5].
To powiedziawszy, screening in silico ma swoje ograniczenia. Nie uwzględnia dobrze stanu chromatyny, cyklu komórkowego ani kontekstu specyficznego dla komórki [1][6][4]. Traktuj to jako filtr, a nie dowód.Zawęża pole, ale nie zastępuje eksperymentalnego potwierdzenia.
Przenieś przewidywane miejsca o najwyższym ryzyku do planu edycji i walidacji.
sbb-itb-ffee270
Lista kontrolna edycji: wybór edytora, dostarczenie i ekspozycja
Używaj edytorów o wysokiej specyficzności i dobrze ocenianych przewodników RNA
Zacznij od przewidywanej listy skróconej miejsc poza celem i użyj jej do wyboru edytora. W większości przypadków, wariant SpCas9 o wysokiej wierności - SpCas9-HiFi, eSpCas9 lub SpCas9-HF1 - jest lepszym domyślnym wyborem niż dziki typ SpCas9 [6][1] . Dziki typ SpCas9 może tolerować do trzech do pięciu niedopasowań par zasad, szczególnie w regionie PAM-dystalnym, co stwarza znaczące ryzyko poza celem w wrażliwych liniach komórkowych [3].
Prosta zasada pomaga tutaj: używaj najmniej aktywnego edytora wysokiej wierności, który nadal dostarcza zamierzony efekt.
Dla edytorów bazowych śledź edytowanie przypadkowe i efekty poza celem RNA oddzielnie od ryzyka poza celem DNA [1] [8] . Są to różne tryby awarii i wymagają oddzielnych kontroli. Jeśli możesz dokonać edycji bez przerw dwuniciowych, edytowanie bazowe lub edytowanie pierwotne może lepiej pasować do procesów o wyższym ryzyku [1][8].
Gdy edytor zostanie wybrany, następnym zadaniem jest utrzymanie jego czasu wewnątrz komórki tak krótko, jak to możliwe.
Ogranicz trwałość edytora poprzez przejściowe dostarczanie i minimalną skuteczną dawkę
Trwałość edytora jest tak samo ważna jak wybór edytora.Im dłużej edytor pozostaje aktywny w komórce, tym więcej czasu ma na działanie w miejscach o niskim prawdopodobieństwie. To sprawia, że format dostarczania staje się głównym punktem kontrolnym.
Używaj przejściowego dostarczania, takiego jak RNPs lub mRNA, i unikaj plazmidowego DNA lub wektorów wirusowych, które wydłużają ekspresję edytora [1] [5] . W praktyce, dostarczanie RNP powinno być domyślne [6].
Dawka również ma znaczenie. Wysokie stężenie nukleazy zwiększa szansę na cięcie w miejscach o niskiej czułości poza celem [5]. Używaj minimalnej skutecznej dawki. Jeśli wydajność transfekcji jest niska, nie dodawaj po prostu więcej odczynnika z nadzieją na najlepsze. To często przesuwa problem zamiast go rozwiązać.
Dodaj zabezpieczenia precyzyjne dla bardziej ryzykownych przepływów pracy
Niektóre przepływy pracy wymagają dodatkowych zabezpieczeń. Dotyczy to szczególnie celów w pobliżu onkogenów, genów supresorowych nowotworów, lub w liniach komórkowych wrażliwych na p53, gdzie jedno zdarzenie poza celem może mieć nieproporcjonalnie wysokie koszty [1][6][3].
Przydatne zabezpieczenia obejmują:
- Sparowane nikazy, które wymagają dwóch pobliskich cięć. Pojedyncze cięcie poza celem jest zazwyczaj naprawiane bez mutacji, więc ryzyko poza celem znacznie spada w porównaniu ze standardowym ustawieniem nukleazy [4][1].
- Indukowalne, kontrolowane światłem lub systemy split-Cas9, które pomagają utrzymać aktywność edytora w wąskim oknie czasowym, gdy dostarczanie jest efektywne, a ekspozycja musi być krótka [1].
- Białka Anti-CRISPR (Acr), które działają jako wyłącznik. Te naturalnie występujące białka Acr mogą dezaktywować kompleks CRISPR-Cas po określonym czasie, dając molekularny hamulec na aktywność edytora [1].
Lista kontrolna po edycji: wykrywanie zdarzeń poza celem i walidacja klonów
Przesiewaj przewidywane miejsca poza celem za pomocą ukierunkowanego sekwencjonowania
Po zakończeniu edycji, najpierw potwierdź zamierzoną zmianę w miejscu docelowym. Dla szybkiego pierwszego przeglądu w komórkach zbiorczych, możesz użyć testu rozszczepienia niedopasowania, takiego jak T7 Endonuclease I, trawienie restrykcyjne lub PCR flankujący.Po prostu bądź ostrożny z interpretacją: każda z tych metod ma swoje ograniczenia czułości, szczególnie w przypadku rzadkich edycji lub wariantów homozygotycznych [9].
Do walidacji na poziomie klonów, ukierunkowane NGS ampliconów jest standardem. Daje ilościowy obraz częstości alleli i może wykrywać warianty na poziomie 0,01% do 0,1% [3].
Sekwencjonuj każde przewidywane miejsce poza celem za pomocą ukierunkowanego NGS ampliconów. To powinien być domyślny krok walidacji.
Przejdź do badań genomowych lub strukturalnych, gdy ryzyko projektu jest wyższe
Przesiewanie miejsce po miejscu nie zawsze wystarcza. Jeśli edytor, docelowe miejsce lub linia komórkowa sugerują ukryte ryzyko, przejdź do badań, które mogą wykryć zdarzenia, których nie przewidziałeś wcześniej.
Badania odkrywcze na poziomie genomu takie jak GUIDE-seq i CIRCLE-seq nie wymagają wcześniejszych list miejsc poza celem.GUIDE-seq może wykrywać miejsca poza celem z częstością indeli tak niską jak 0,03% [2]. CIRCLE-seq może identyfikować do 94% miejsc poza celem in vitro [3]. Te metody są przydatne, gdy kontekst typu komórki może maskować aktywność poza celem.
Jeśli martwisz się o duże rearanżacje, standardowe odczyty ampliconów mogą pominąć główny problem. Delecje, inwersje i translokacje wymagają testów zbudowanych dla zmian strukturalnych, takich jak CAST-seq i UDiTaS [1].
Sekwencjonowanie całego genomu (WGS) jest najszerszą opcją. Może wykrywać indeli, zmiany strukturalne i zmiany liczby kopii w całym genomie [1]. Kompromis to głębokość i koszt: zazwyczaj wymaga 20–60× pokrycia, co czyni go nieodpowiednim do rutynowego badania przesiewowego dużych populacji [1].
Użyj NGS ukierunkowanego na amplicony dla przewidywanych miejsc. Przejdź do badań genomowych lub strukturalnych dla projektów o wyższym ryzyku. Dla edytorów bazowych lub pierwotnych dodaj RNA-seq, aby sprawdzić efekty poza celem na poziomie RNA.
Wybierz wiele niezależnych klonów i udokumentuj kryteria uwalniania
Po sprawdzeniu sekwencji przetestuj fenotyp w więcej niż jednym klonie.
Nie kontynuuj z jednym edytowanym klonem. Izoluj i rozszerz co najmniej dwie do trzech niezależnych populacji klonalnych i porównaj je z nieedytowaną kontrolą rodzicielską [4][9]. Usuń klony wykazujące niestabilność lub dryf fenotypowy [3]. Następnie potwierdź zamierzony edytowany stan allelu, czy jest heterozygotyczny czy homozygotyczny, używając ukierunkowanego ampliconu NGS [9].
Dokumentacja nie jest pracą administracyjną na końcu. Jest częścią wydania klonu. Zarejestruj tło linii rodzicielskiej, projekt sgRNA, wariant nukleazy, metodę dostarczania oraz wszystkie wyniki QC [3]. Klon powinien przejść dalej tylko wtedy, gdy zamierzony edytowany stan jest potwierdzony, przewidywane miejsca poza celem są jasne, a pełna dokumentacja jest na miejscu.
Edytowanie genomu za pomocą CRISPR: Jak skutecznie minimalizować efekty poza celem
Wniosek: lista kontrolna w trzech fazach dla czystszych edycji linii komórkowych
Podsumowując, lista kontrolna traktuje kontrolę poza celem jako proces etapowy, a nie jednorazową kontrolę QC. Cel jest prosty: zredukować ryzyko wcześnie, ograniczyć aktywność edytora, a następnie zweryfikować wynik.
Głębokość walidacji powinna odpowiadać ryzyku. Uwalniaj tylko wiele niezależnych klonów potwierdzonych w zamierzonym stanie allelu.
Najczęściej zadawane pytania
Dlaczego nie polegać na jednym klonie?
Poleganie na jednym klonie jest ryzykowne. Edytowanie CRISPR nie jest idealnie specyficzne, więc może wprowadzać niezamierzone mutacje poza celem.
Dlatego zespoły zazwyczaj rozszerzają wiele populacji klonalnych. Dzięki temu łatwiej jest znaleźć linię, która posiada zamierzoną edycję na celu bez szkodliwych zmian poza celem.
Jest jeszcze jeden powód: linie komórkowe mogą wykazywać heterogeniczność genetyczną. Sekwencjonowanie wielu klonów pomaga potwierdzić, że zamierzony nokaut homozygotyczny lub inna modyfikacja miejsca docelowego jest obecna w całym loci docelowym.
Kiedy amplicon NGS jest wystarczający?
Sekwencjonowanie nowej generacji oparte na ampliconach jest często wystarczające, gdy potrzebujesz ukierunkowanego i opłacalnego sposobu potwierdzenia potencjalnych miejsc poza celem wskazanych przez narzędzia obliczeniowe lub inne metody przesiewowe.
Całogenomowe sekwencjonowanie jest nadal jedynym sposobem na pełne określenie efektów poza celem. Jednak dla wielu zastosowań taki poziom analizy po prostu nie jest potrzebny.
Jak wybrać najbezpieczniejszy edytor?
Wybierz najmniej aktywny wariant nukleazy CRISPR, który nadal dobrze przecina twoje miejsce docelowe.
Nie możesz wybrać najlepszego wariantu tylko na podstawie przewidywań. Jedynym niezawodnym sposobem jest przeprowadzenie małego przesiewu wśród wybranych wariantów nukleazy i odczytanie edycji za pomocą sekwencjonowania nowej generacji.
Dla hodowanego mięsa R&D, to daje praktyczną ścieżkę naprzód: zacznij od krótkiej listy wariantów, a następnie testuj słabsze krok po kroku, aż znajdziesz najmniej aktywną opcję, która nadal efektywnie edytuje docelowe miejsce.









