

Rejestry partii są kluczowe dla zgodności i bezpieczeństwa produktów. Dokumentują każdy etap produkcji, zapewniając spełnienie standardów regulacyjnych. Dla producentów mięsa hodowlanego, utrzymanie sterylności i szczegółowych rejestrów jest niepodważalne. Inspekcje FDA często podkreślają problemy takie jak brakujące dane, niekompletne przeglądy i słabe działania naprawcze, co może prowadzić do ostrzeżeń lub zakłóceń.
Kluczowe Wnioski:
- Rejestry Partii: Dwa typy - Główny Rejestr Partii (MBR) (przepis) i Rejestr Produkcji Partii (BPR) (wykonanie).
- Typowe Problemy: Błędy ludzkie (50% problemów), brakujące kontrole w trakcie procesu, niekompletne przeglądy i słabe systemy CAPA (Działania Korygujące i Zapobiegawcze).
- Standardy FDA: Przestrzeganie zasad ALCOA+ (Przypisywalne, Czytelne, Współczesne, Oryginalne, Dokładne, Kompletne, Spójne, Trwałe, Dostępne) jest obowiązkowe.
- Rozwiązania: Niezależne audyty, elektroniczne rejestry partii i rygorystyczna weryfikacja dostawców mogą zminimalizować błędy i poprawić zgodność.
Firmy zajmujące się mięsem hodowlanym, takie jak UPSIDE Foods, ustanowiły punkt odniesienia, zapewniając szczegółową dokumentację, śledzenie wejść i szybkie działania naprawcze. Ucząc się z tych praktyk, producenci mogą unikać pułapek regulacyjnych i utrzymywać wysokie standardy jakości.
Kompleksowy przewodnik po dokumentacji i prowadzeniu rejestrów dla zgodności z FDA w naukach przyrodniczych
sbb-itb-ffee270
Typowe problemy w dokumentacji rejestrów partii
Raporty z inspekcji FDA konsekwentnie podkreślają powtarzający się problem: odchylenia w przeglądzie rejestrów produkcji należą do najczęściej cytowanych przez regulatorów braków GMP [7]. Dla producentów mięsa hodowlanego te niedociągnięcia wykraczają poza zwykłe błędy administracyjne - zagrażają zdolności do wykazania utrzymania sterylnych warunków. Problemy te pojawiają się w różnych formach, jak pokazano w poniższych przykładach.
Niekompletne przeglądy i niezgodności
Jednym z powszechnych problemów jest brak dokładnego przeglądu zapisów partii przez jednostki kontroli jakości. Zamiast być integralną częścią procesu wydania, przeglądy często odbywają się reaktywnie - dopiero po pojawieniu się problemu z produktem [7]. Takie podejście pozostawia znaczące luki w zapisach produkcyjnych.
Na przykład, Davis City Pharmacy otrzymała obserwację FDA 483 z powodu brakujących w zapisach partii kluczowych szczegółów, takich jak ilości składników, kroki operacyjne i inicjały personelu. Podobnie, CAPS została zacytowana za brak wymaganych podpisów i weryfikacji przeglądających w kluczowych wpisach [3]. Te uchybienia nie są odosobnionymi przypadkami; badania pokazują, że około 52% naruszeń dokumentacji eskaluje, gdy brakuje solidnych systemów zarządzania bioprocesami [3].
"To nie złożoność procesu wywołuje cytacje - to niespójność, niekompletność i słaby nadzór." - GXP Auditing & Consulting Services [5]
Brakujące zapisy kontroli w trakcie procesu
Kolejnym częstym niedociągnięciem jest brak odpowiedniej dokumentacji dla kontroli w trakcie procesu. Te zapisy są kluczowe, zwłaszcza w krytycznych punktach kontrolnych w operacjach aseptycznych. Na przykład, Nephron Sterile Compounding Center otrzymało cytacje za brak dokumentacji niezbędnych kroków ubierania i procedur aseptycznych w ich zapisach partii [3]. Dla producentów mięsa hodowlanego, gdzie sterylność jest kluczowa, takie pominięcia uniemożliwiają potwierdzenie zgodności z środkami kontroli zanieczyszczeń.
Amphastar został również oznaczony za brak dochodzenia lub dokumentowania nieoczekiwanych odchyleń wydajności lub rozbieżności w produkcji [3]. Ryzyka takich przeoczeń są wyraźne. W jednym przypadku, niezidentyfikowany zakład farmaceutyczny w latach 2024/2025 przechowywał ukończone zapisy partii na otwartych półkach i biurkach. Śledczy FDA odkryli brakujące strony, w tym siedem z jednego zapisu, oraz brak całej sekcji "Rozwiązanie Syntezy" w innym [6].
Niepowodzenia w zakresie CAPA i weryfikacji GMP dostawców
Poza błędami w dokumentacji, brak skutecznych procesów korygujących i weryfikacji dostawców dodatkowo podważa wiarygodność zapisów partii.Gdy występują odchylenia produkcyjne bez odpowiadającego im raportu Działań Korygujących i Zapobiegawczych (CAPA), integralność zapisów partii jest zagrożona [7]. Na przykład, Eugia Pharma Specialities Limited, kontrolowana między 22 stycznia a 2 lutego 2024 roku, otrzymała FDA 483 za niewystarczające przeglądanie rozbieżności. Ich nieskuteczny system CAPA i niekompletne dochodzenia doprowadziły do powtarzających się problemów produkcyjnych, zmuszając do całkowitej przebudowy procedur dochodzeniowych i CAPA [9].
Podobnie, podczas inspekcji od 26 września do 25 października 2023 roku, Stokes Healthcare Inc. wykazała słabe zarządzanie rozbieżnościami. Firma nie rozszerzyła dochodzeń na wszystkie dotknięte partie i opóźniła zakończenie swoich analiz [9].
"Rozbieżność bez odpowiadającego raportu CAPA lub odchylenia? To jest naruszenie zgodności." - GXP Auditing & Usługi doradcze [5]
Problemy z weryfikacją dostawców dodają kolejny poziom złożoności. Empower Clinic Services LLC została wymieniona podczas inspekcji od 18 lipca do 5 sierpnia 2022 roku za niewystarczające procedury kontroli jakości, w tym niewystarczające kwalifikacje dostawców i słabe procesy dochodzeniowe [9]. Dla producentów mięsa hodowlanego, którzy polegają na pożywkach wzrostowych, liniach komórkowych i innych kluczowych składnikach, zapewnienie zgodności dostawców z GMP jest kluczowe dla utrzymania integralności zapisów partii.
Wymagania FDA dotyczące zapisów partii
Zasady FDA dotyczące zapisów partii koncentrują się wokół 21 CFR Część 117, które ustanawiają podstawowe zasady bezpieczeństwa żywności.Jeśli chodzi o mięso hodowlane, gdzie utrzymanie sterylności podczas fazy hodowli komórek jest kluczowe, dokumentacja często musi spełniać surowsze standardy Części 111 lub Części 211, oprócz Części 117 [10][14]. To podkreśla, jak precyzyjna dokumentacja jest niezbędna dla zapewnienia bezpieczeństwa i skuteczności produkcji mięsa hodowlanego.
Podstawowe standardy dla zapisów partii
Każda partia wymaga dwóch kluczowych dokumentów:
- Główny zapis partii (MBR): Zaakceptowany szablon opisujący proces produkcji.
- Zapis produkcji partii (BPR): Szczegółowy zapis tego, co faktycznie dzieje się podczas przebiegu produkcji [12][2].
BPR musi zawierać szczegóły takie jak numery partii lub serii, dane dotyczące sprzętu, daty czyszczenia, identyfikatory komponentów, dokładne pomiary oraz porównania rzeczywistych i teoretycznych wydajności [10][14].
"Rejestr produkcji partii musi dokładnie podążać za odpowiednim głównym rejestrem produkcji i każdy krok w produkcji partii musi być wykonany." – 21 CFR 111.255 [12]
Każdy krytyczny krok musi być zapisany natychmiast, z inicjałami zarówno wykonawcy, jak i weryfikatora [10][11]. FDA wymaga przestrzegania zasad ALCOA(+), co oznacza, że zapisy muszą być Przypisane, Czytelne, Współczesne, Oryginalne i Dokładne - a także Kompletne, Spójne, Trwałe i Dostępne [1].
Jeśli wystąpi jakiekolwiek odstępstwo od Głównego Rejestru Produkcji, musi zostać dokładnie zbadane. Obejmuje to dokumentowanie problemu, przeprowadzenie analizy przyczyn źródłowych oraz wdrożenie planu działań korygujących i zapobiegawczych (CAPA) [8] [1]. Początkowe oceny odstępstw powinny być rejestrowane w ciągu 24–48 godzin od wykrycia [8]. Dla zakładów korzystających z systemów elektronicznych, zgodność z 21 CFR Part 11 jest obowiązkowa. Obejmuje to zweryfikowane podpisy elektroniczne i bezpieczne, oznaczone czasem ścieżki audytu [8] [1].
Procedury przechowywania i przeglądu dokumentacji
Właściwe procesy przechowywania i przeglądu dokumentacji są kluczowe dla zachowania zgodności i zapewnienia bezpieczeństwa produktu.W produkcji sterylnej, takiej jak mięso hodowlane, każdy szczegół w zapisach partii musi przejść skrupulatną kontrolę. Zespół Kontroli Jakości (QC) jest odpowiedzialny za przegląd wszystkich zapisów partii, monitorowanie wyników i testowanie danych przed zatwierdzeniem partii do dystrybucji [10] [13].
"Wszystkie zapisy dotyczące produkcji i kontroli produktów leczniczych muszą być przeglądane i zatwierdzane przez jednostkę kontroli jakości przed wypuszczeniem lub dystrybucją partii." – 21 CFR 211.192 [2]
Producenci często dążą do zakończenia 95% przeglądów partii w ciągu 30 dni od produkcji [2] . Jednak w przypadku bardziej złożonych procesów sterylnych związanych z mięsem hodowlanym, przeglądy zazwyczaj trwają 7–10 dni, a wysokowydajne zakłady osiągają czasy poniżej 7 dni [2]. Systemy elektronicznych rejestrów partii mogą znacznie przyspieszyć te przeglądy, takie jak te zintegrowane z systemami produkcji mięsa hodowanego, - skracając czas o połowę w porównaniu z metodami papierowymi - pod warunkiem, że są one zwalidowane do spełnienia wymagań Części 11 i utrzymania integralności danych [1].
Co Zrobiły Dobrze Firmy Zatwierdzone Przez FDA Produkujące Mięso Hodowane
Firmy produkujące mięso hodowane zatwierdzone przez FDA ustawiły poprzeczkę wysoko, przyjmując praktyki, które rozwiązują wyzwania związane z dokumentacją i spełniają rygorystyczne standardy bezpieczeństwa.
Kiedy UPSIDE Foods stało się pierwszą firmą produkującą mięso hodowane, która przeszła konsultację przedrynkową FDA w listopadzie 2022 roku, ustanowili model dla branży.FDA wydała list "bez dalszych pytań" po dokładnym przeglądzie ich procesu produkcyjnego, który obejmował ustanowienie linii komórkowych, banki komórek, kontrole produkcji oraz wszystkie komponenty i wkłady [16]. To osiągnięcie podkreśliło znaczenie szczegółowej dokumentacji w spełnianiu surowych wymagań FDA.
Spełnianie standardów sterylności i zgodności
Wyjątkowym osiągnięciem UPSIDE Foods było ich wyczerpujące podejście do śledzenia wkładów. Każdy komponent produkcji był starannie udokumentowany, zapewniając jasny łańcuch odpowiedzialności od początkowej linii komórkowej do finalnego produktu [16]. Ten poziom przejrzystości pozwolił recenzentom FDA prześledzić każdy krok procesu produkcyjnego, potwierdzając, że wszystkie standardy bezpieczeństwa były konsekwentnie spełniane.
"Konsultacja przedrynkowa FDA z firmą obejmowała ocenę procesu produkcyjnego firmy oraz materiału komórkowego hodowanego w procesie produkcyjnym, w tym ustanowienie pierwotnych i unieśmiertelnionych linii komórkowych oraz banków komórek, kontroli produkcji i wszystkich komponentów i wkładów." – U.S. Food and Drug Administration [16]
Inne odnoszące sukcesy firmy poszły w ich ślady, wdrażając szczegółową dokumentację procesów aseptycznych. Obejmowało to krytyczne kroki, takie jak procedury ubierania się i operacje sterylnego obchodzenia się [3]. W przeciwieństwie do wcześniejszych niepowodzeń w dokumentacji, te firmy stosowały systemy przeglądów warstwowych, obejmujące kontrole operatorów, nadzór produkcji i przeglądy jednostek jakości, aby wychwycić potencjalne błędy przed wydaniem partii [15]. Systemy elektronicznych rejestrów partii również odgrywały kluczową rolę, wymuszając obowiązkowe zatwierdzenia na każdym etapie i utrzymując niezmienne ścieżki audytu zgodnie z wymaganiami 21 CFR Część 11 [3][2].
Te rygorystyczne praktyki naturalnie rozszerzały się na sposób, w jaki firmy radziły sobie z odchyleniami i niepowodzeniami.
Procesy CAPA dla niepowodzeń partii
Kiedy partie nie spełniały specyfikacji, firmy zatwierdzone przez FDA podejmowały szybkie i systematyczne działania. Ich procesy Działań Korygujących i Zapobiegawczych (CAPA) obejmowały formalne analizy przyczyn źródłowych, oceny wpływu oraz jasno udokumentowane działania korygujące [3]. Wszelkie odchylenia były zarządzane w ramach zintegrowanego systemu zapewnienia jakości, zapewniając, że wszystkie problemy były dokładnie badane, uzasadniane i dokumentowane przed kontynuacją produkcji [2].
Patrząc w przyszłość, integralność danych ma być głównym celem działań egzekucyjnych FDA na lata 2024–2025 [1].
Jak poprawić praktyki dotyczące rejestrów partii
Wzmocnienie praktyk dotyczących rejestrów partii wymaga precyzyjnej dokumentacji, aby rozwiązać typowe problemy często identyfikowane podczas inspekcji FDA. Oto kilka strategii, które pomogą sprostać kluczowym wyzwaniom.
Przeprowadzaj niezależne audyty rejestrów partii
Regularne audyty zewnętrzne mogą ujawnić problemy, które mogą zostać przeoczone podczas wewnętrznych przeglądów. Zacznij od skupienia się na krytycznych systemach, takich jak Systemy Zarządzania Informacjami Laboratoryjnymi (LIMS), Systemy Wykonawcze Produkcji (MES) i Planowanie Zasobów Przedsiębiorstwa (ERP). Priorytetowo traktuj dokumenty o dużym wpływie regulacyjnym, takie jak zapisy testów uwalniania, dane stabilności i rejestry produkcji partii.
Jedną z efektywnych metod jest testowanie pobierania próbek.Losowo wybierz ostatnie partie i odtwórz ich historię produkcji oraz badań laboratoryjnych. Może to pomóc w zidentyfikowaniu brakujących danych, niekompletnych podpisów lub luk w dokumentacji, które mogą prowadzić do cytatów regulacyjnych. Porównaj systemowo generowane ścieżki audytu z ręcznymi wpisami, aby zidentyfikować nieautoryzowane zmiany lub usunięcia.
Przejrzyj wszystkie raporty Out-of-Specification (OOS) i Out-of-Trend (OOT) z ostatniego roku. Oceń, czy analizy przyczyn źródłowych były dokładne i czy Działania Korygujące i Zapobiegawcze (CAPA) zostały odpowiednio wdrożone. Warto zauważyć, że problemy z dokumentacją stanowią 21% ostrzeżeń FDA, podczas gdy błędy ludzkie przyczyniają się do 50% problemów z zapisami partii w produkcji farmaceutycznej [2].
"To nie złożoność procesu wywołuje cytaty - to niespójność, niekompletność i słaby nadzór." – GXP Auditing & Consulting Services [5]
Symuluj inspekcje regulacyjne poprzez okresowe przeglądy próbne. Ta praktyka pomaga zespołom rozpoznać niespójności i potencjalne problemy z integralnością danych przed faktycznym audytem. Upewnij się, że wszystkie zapisy spełniają zasady ALCOA+: Przypisywalne, Czytelne, Współczesne, Oryginalne, Dokładne, Kompleksowe, Spójne, Trwałe i Dostępne.
Gdy integralność dokumentacji jest solidna, skoncentruj się na weryfikacji jakości wszystkich surowców produkcyjnych.
Testuj Wszystkie Surowce pod kątem Zanieczyszczeń Mikrobiologicznych
Niezależne testy jałowości i mocy dla wszystkich surowców są niezbędne - nie polegaj wyłącznie na Certyfikatach Analizy (CoA) dostawców. Jest to szczególnie istotne dla producentów mięsa hodowlanego, ponieważ zanieczyszczenie może zagrozić całym partiom.
Na przykład, w lutym 2013 roku, Central Admixture Pharmacy Services otrzymało upomnienia od FDA z powodu niewystarczającej kontroli mikrobiologicznej podczas zwalniania partii produktów sterylnych. Firma musiała wprowadzić szczegółowe procedury kontroli mikrobiologicznej do swoich Standardowych Procedur Operacyjnych (SOPs) [4].
Kontrolne punkty mikrobiologiczne w trakcie procesu mogą zapobiec nadmiernemu poleganiu na testach końcowych produktów. Włącz te punkty kontrolne do SOPs zwalniania partii i utrzymuj ścisłą bieżącą dokumentację. Zapisuj wszystkie wyniki testów i kroki produkcyjne na bieżąco, aby uniknąć wstecznego datowania lub opóźnionych wpisów, co mogłoby skutkować upomnieniami od FDA.
Utrzymuj kompleksowe pliki dostawców, w tym CoAs, raporty z audytów, umowy jakościowe oraz historię wszelkich odchyleń związanych z materiałami przychodzącymi.
Wzmocnienie praktyk dotyczących dokumentacji partii dodatkowo obejmuje dostosowanie procesów do ustalonych standardów, takich jak HACCP i GCCP.
Wyrównaj dokumentację z normami HACCP i GCCP
Włączenie zasad Analizy Zagrożeń i Krytycznych Punktów Kontroli (HACCP) do dokumentacji partii zapewnia monitorowanie i dokumentowanie krytycznych zmiennych procesowych w całym procesie produkcji. Obejmuje to ustanowienie punktów kontrolnych testów mikrobiologicznych w trakcie procesu, zamiast polegania wyłącznie na testach końcowych.
Dla producentów mięsa hodowlanego przestrzeganie standardów Dobrej Praktyki Hodowli Komórek (GCCP) jest równie istotne. Dokumentacja partii powinna zawierać szczegóły dotyczące manipulacji aseptycznych, procedur ubierania się oraz monitorowania środowiska powiązanego z kryteriami zwolnienia partii [3][4]. Te kroki pomagają utrzymać zgodność i zapewnić bezpieczeństwo produktu.
Dane branżowe pokazują, że 52% naruszeń dokumentacji eskaluje, gdy nie ma odpowiedniego oprogramowania do zarządzania produkcją partii [3][4]. Przykład: w lutym 2023 roku Nephron Sterile Compounding Centre otrzymało obserwację od FDA z powodu braku procedur kontrolnych weryfikujących krytyczne zmienne procesu przed wydaniem partii [4]. To podkreśla potrzebę proaktywnej dokumentacji zgodnej z uznanymi standardami.
Przejście na Elektroniczne Rejestry Partii (EBR) może znacząco zmniejszyć błędy w dokumentacji - nawet o 50% - dzięki zbieraniu danych w czasie rzeczywistym i zautomatyzowanym przepływom pracy [2]. Systemy te sygnalizują brakujące wyniki testów mikrobiologicznych lub niekompletne przeglądy przed postępem partii, minimalizując błędy ludzkie.
"FDA oczekuje, że zapisy będą ALCOA(+): Przypisywalne, Czytelne, Współczesne, Oryginalne, Dokładne - plus Kompletne, Spójne, Trwałe i Dostępne." – Atlas Compliance [1]
Każda niewyjaśniona rozbieżność lub odchylenie w zapisach partii powinna być powiązana z formalnym dochodzeniem i systemem CAPA. Ogranicz uprawnienia do zapisu i usuwania, aby chronić integralność elektronicznych danych z testów mikrobiologicznych. Konkurencyjni producenci dążą do przeglądu i wydania 95% partii w ciągu 30 dni od zakończenia produkcji [2].
Te działania nie tylko zmniejszają ryzyko cytacji, ale także są zgodne z rygorystycznymi standardami dokumentacji podkreślonymi w ostatnich inspekcjach FDA.
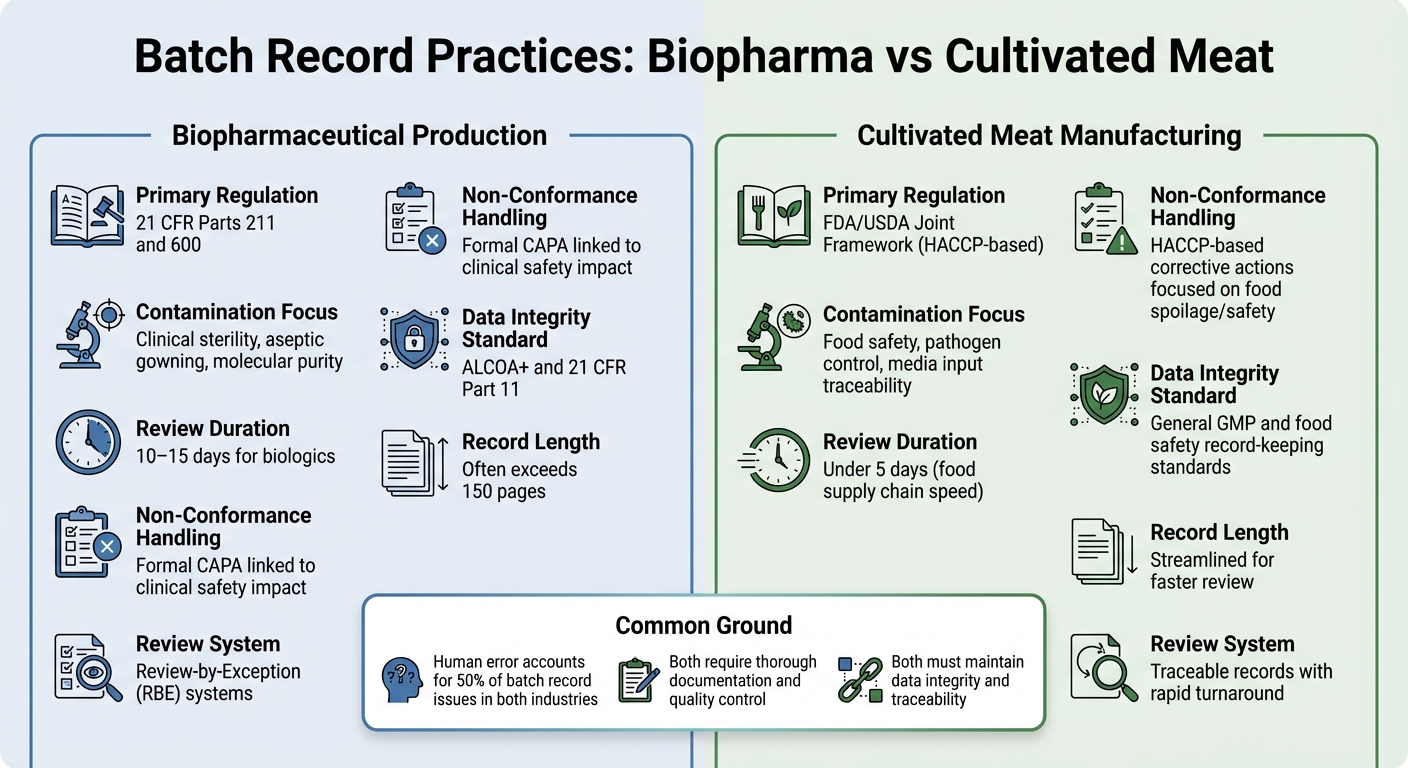
Biopharma vs Mięso Hodowlane: Różnice w Dokumentacji Partii
Porównanie Wymagań Dokumentacji Partii w Biopharmaceutykach i Mięsie Hodowlanym
Analiza różnic w praktykach dokumentacji partii między produkcją biopharmaceutyków a produkcją mięsa hodowlanego oferuje wyraźniejszy obraz tego, jak wymagania regulacyjne kształtują priorytety dokumentacyjne w tych branżach.
Oba sektory wymagają dokładnej dokumentacji, ale ich ramy regulacyjne i cele kontrolne różnią się znacznie. W biopharmaceutykach dokumentacja partii jest ściśle regulowana zgodnie z 21 CFR Części 211 i 600, które wymagają, aby jednostka Kontroli Jakości przeglądała i zatwierdzała wszystkie zapisy produkcji i kontroli przed wypuszczeniem partii [2]. Producenci mięsa hodowlanego, z drugiej strony, zazwyczaj stosują standardy HACCP i GCCP.Te koncentrują się bardziej na bezpieczeństwie żywności i kontroli patogenów, a nie na sterylności klinicznej wymaganej dla biologicznych preparatów do wstrzykiwań.
Rekordy partii w biotechnologii farmaceutycznej są często obszerne, czasami przekraczają 150 stron, a proces przeglądu może trwać 10–15 dni. Aby to usprawnić, wiele firm biotechnologicznych używa systemów Review-by-Exception (RBE), które podsumowują kluczowe odchylenia w zwięzłym raporcie. Tymczasem producenci mięsa hodowlanego dążą do uzyskania śledzonych rekordów, które można przeglądać w mniej niż pięć dni, co odzwierciedla szybsze tempo łańcucha dostaw żywności [2].
Zawartość tych rekordów również podkreśla różne priorytety. Inspekcje biotechnologii farmaceutycznej często koncentrują się na szczegółach dotyczących aseptycznego przetwarzania, takich jak procedury ubierania się i kontrole środowiskowe. Natomiast rekordy mięsa hodowlanego muszą kłaść nacisk na wkłady do mediów i testy mikrobiologiczne, aby zapewnić bezpieczeństwo żywności.Dla mięsa hodowlanego wyzwaniem jest śledzenie złożonych składników pożywki, dokumentowanie testów mikrobiologicznych dla wszystkich materiałów oraz spełnianie krytycznych limitów bezpieczeństwa żywności - bez przestrzegania bardziej rygorystycznych wymagań dotyczących sterylności w farmacji.
Trendy zanieczyszczeń i niezgodności
| Funkcja | Produkcja biofarmaceutyczna | Produkcja mięsa hodowlanego |
|---|---|---|
| Podstawowe przepisy | 21 CFR Części 211 i 600[2] | Wspólne ramy FDA/USDA (oparte na HACCP) |
| Skupienie na zanieczyszczeniach | Jałowość kliniczna, aseptyczne ubieranie, czystość molekularna[2] | Bezpieczeństwo żywności, kontrola patogenów, śledzenie wejść do mediów |
| Czas przeglądu | 10–15 dni dla produktów biologicznych[2] | Poniżej 5 dni (szybkość łańcucha dostaw żywności) |
| Obsługa niezgodności | Formalne CAPA powiązane z wpływem na bezpieczeństwo kliniczne [2] | Działania korygujące oparte na HACCP skoncentrowane na psuciu się/bezpieczeństwie żywności |
| Standard integralności danych | ALCOA+ i 21 CFR Część 11 [1] | Ogólne standardy prowadzenia dokumentacji GMP i bezpieczeństwa żywności |
Podczas gdy wskaźniki błędów ludzkich są podobne w obu branżach - około 50% problemów z zapisami partii wynika z błędów ludzkich [2] - stawki są różne.W biopharmacji nawet pojedyncze nieudokumentowane odstępstwo może mieć poważne konsekwencje dla bezpieczeństwa pacjentów. W przypadku mięsa hodowlanego ryzyko zanieczyszczenia dotyczy bardziej patogenów przenoszonych przez żywność i psucia się, co może wpłynąć na całe partie produkcyjne.
Wniosek
Rejestry partii służą jako oficjalny dziennik dla każdej partii produkcji mięsa hodowlanego - jeśli krok nie jest zarejestrowany, organy regulacyjne uznają go za nieprzeprowadzony [6][3]. To podkreśla znaczenie precyzyjnej dokumentacji i ścisłej kontroli jakości.
Inspekcje FDA podkreślają, że integralność danych musi być zgodna z zasadami ALCOA+ [1]. Zespoły Kontroli Jakości są zobowiązane do przeglądu i zatwierdzenia wszystkich zapisów produkcyjnych przed wypuszczeniem partii [2][17], a wszelkie odstępstwa muszą być niezwłocznie zbadane z udokumentowaną analizą przyczyn źródłowych [2][5]. Podczas gdy błąd ludzki stanowi 50% problemów z zapisami partii, przeglądy na dwóch poziomach i ustrukturyzowane procesy CAPA (Działania Korygujące i Zapobiegawcze) mogą pomóc w zmniejszeniu tych ryzyk [2][5].
"To nie złożoność procesu wywołuje cytaty - to niespójność, niekompletność i słaby nadzór." - GXP Auditing & Consulting Services [5]
Aby pokonać te wyzwania, producenci mięsa hodowlanego powinni skupić się na niezależnych audytach, rygorystycznym testowaniu składników bezpiecznych dla żywności pod kątem zanieczyszczeń mikrobiologicznych oraz zapewnieniu, że dokumentacja jest zgodna ze standardami HACCP i GCCP. Wdrożenie elektronicznych systemów rejestracji partii, zwalidowanych zgodnie z 21 CFR Część 11 [1], może znacznie zminimalizować błędy i przyspieszyć procesy przeglądu.
Środowisko regulacyjne wymaga precyzji, ale jest możliwe do nawigacji.Ucząc się na błędach biotechnologii - takich jak brakujące podpisy w Qinhuangdao Zizhu Pharmaceutical [17], niewystarczająca podwójna weryfikacja w Terumo Corp [18] , oraz niewłaściwa dokumentacja odchyleń w Torrent Pharmaceuticals [18] - firmy zajmujące się mięsem hodowlanym mogą od początku ustanowić zgodne systemy. Włączenie tych lekcji umożliwia proaktywną zgodność i spójną jakość. Bezpieczne przechowywanie dokumentacji, terminowe raportowanie odchyleń i przeprowadzanie realistycznych audytów próbnych zapewni, że zapisy partii będą gotowe do inspekcji, a serie produkcyjne w pełni śledzone.
Aby uzyskać więcej zasobów i fachowych wskazówek dotyczących utrzymania wysokich standardów produkcji w produkcji mięsa hodowlanego, odwiedź
FAQs
Co powinien zawierać zapis partii dla mięsa hodowlanego?
Zapis partii dla mięsa hodowlanego służy jako kompleksowy dziennik całego procesu produkcyjnego. Musi zawierać szczegółowe instrukcje przetwarzania, zapisy wykonania krok po kroku, i zanotować wszelkie odchylenia, które występują podczas produkcji. Dodatkowo powinien dokumentować testy w trakcie procesu oraz testy uwalniania, aby potwierdzić, że produkt spełnia normy bezpieczeństwa, jakości i regulacyjne.
Jak możemy udowodnić jałowość za pomocą zapisów partii?
Udowodnienie jałowości za pomocą zapisów partii polega na dokładnym zbadaniu udokumentowanych procedur sterylizacji, wyników testów i raportów kontroli jakości mediów, aby upewnić się, że spełniają one wymagania regulacyjne.Konieczne jest zajęcie się wszelkimi odchyleniami lub nieudanymi testami poprzez szczegółowe dochodzenia i CAPA (Działania Korygujące i Zapobiegawcze). Ten proces zapewnia, że każdy krok został przestrzegany, a wszelkie problemy zostały odpowiednio rozwiązane, aby utrzymać standardy sterylności.
Kiedy wymagane są elektroniczne zapisy partii (Część 11)?
Elektroniczne zapisy partii są niezbędne zgodnie z Częścią 11, gdy systemy elektroniczne są używane do dokumentowania, badania i uzasadniania odchyleń w zapisach partii. Odgrywają one kluczową rolę w zapewnieniu zgodności z 21 CFR Part 211.192 , ochronie integralności danych, spełnianiu terminów dochodzeń i zapewnieniu skutecznego nadzoru zarządzania.









