

W pomieszczeniach czystych, zgodność z GMP zapewnia spójną jakość i bezpieczeństwo poprzez wymaganie szczegółowego monitorowania i dokładnych zapisów danych. Dla zakładów produkujących mięso hodowlane jest to szczególnie ważne, ponieważ nawet drobne odchylenia w warunkach pomieszczeń czystych mogą zagrozić wzrostowi komórek lub zanieczyścić partie produkcyjne.
Kluczowe wnioski:
- Standardy GMP: Skupiają się na integralności danych, zgodnie z ramami ALCOA+ (Atrybutywność, Czytelność, Współczesność, Oryginalność, Dokładność, Kompletność, Spójność, Trwałość, Dostępność).
- Krytyczne Parametry: Monitoruj cząstki powietrza, liczbę drobnoustrojów, temperaturę, wilgotność i ciśnienie, aby wcześnie wykrywać ryzyka. Wymaga to wyboru precyzyjnych czujników zdolnych do utrzymania tych krytycznych parametrów.
- Systemy Danych: Używaj zwalidowanych systemów kontroli bioprocesów z dostępem opartym na rolach, ścieżkami audytu i bezpiecznym przechowywaniem zarówno dla zapisów elektronicznych, jak i papierowych.
- Typowe ryzyka: Unikaj błędów w ręcznym przetwarzaniu danych, zmian konfiguracji bez kontroli oraz niewłaściwych praktyk przechowywania.
- Dostosowane GMP dla mięsa hodowanego: Dostosuj strategie monitorowania, aby uwzględnić unikalne ryzyka, takie jak warunki w bioreaktorze i pozostałości środków czyszczących.
Dla R&D mięsa hodowanego, solidne zarządzanie danymi zapewnia bezpieczeństwo produktu, zgodność z przepisami i skalowalność operacji. Proaktywne adresowanie znanych podatności pozwala uniknąć kosztownych problemów regulacyjnych w przyszłości.
Kluczowe wymagania GMP dla integralności danych w czystych pomieszczeniach
Zrozumienie zasad ALCOA+
Podstawą integralności danych GMP jest ramy ALCOA+. Organy regulacyjne, takie jak MHRA, EMA, i WHO, używają ich do określenia, czy zapisy z czystych pomieszczeń są godne zaufania.ALCOA+ oznacza: Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available. Każdy z tych terminów ma praktyczne znaczenie w operacjach w pomieszczeniach czystych.
- Attributable: Każdy wpis - czy to liczba cząstek, odczyt ciśnienia, czy dziennik czyszczenia - musi wyraźnie wskazywać, kto go zarejestrował, wraz z datą, godziną i odpowiednimi szczegółami instrumentu.
- Legible: Zapisy muszą być łatwe do odczytania i zrozumienia, zapewniając jasność podczas przeglądów lub inspekcji.
- Contemporaneous: Dane muszą być rejestrowane w czasie rzeczywistym. Opóźnione lub retrospektywne wpisy mogą zagrozić wiarygodności zapisów.
- Original: Dane powinny pozostać w swojej pierwotnej formie, bez nieautoryzowanych edycji lub zmian.
- Dokładne: Zarejestrowane wartości muszą prawdziwie odzwierciedlać zaobserwowane wyniki, wolne od błędów lub manipulacji.
- Kompletne: Wszystkie istotne wpisy, w tym odchylenia lub wyniki poza specyfikacją, muszą być udokumentowane.
- Spójne, Trwałe, i Dostępne: Rekordy powinny być zachowane w odpowiedniej kolejności, przechowywane w nienaruszonym stanie przez wymagany okres przechowywania i łatwo dostępne do przeglądu lub inspekcji.
Regulatorzy kładą duży nacisk na te zasady. Na przykład, przegląd inspekcji MHRA ujawnił, że ponad 80% oświadczeń o niezgodności z GMP w okresie trzech lat dotyczyło problemów z integralnością danych [5]. Aby wdrożyć ALCOA+ w codziennej pracy, obiekty mogą przyjąć dobrze zorganizowane formularze, wymusić obowiązkowe pola i przeprowadzać regularne przeglądy ścieżki audytu.
Z ALCOA+ jako fundamentem, kolejnym krokiem jest zapewnienie, że te zasady są przestrzegane w systemach papierowych, elektronicznych i hybrydowych.
Zapewnienie integralności danych we wszystkich formatach
W zakładach produkujących mięso hodowlane, gdzie dane bezpośrednio wpływają na decyzje dotyczące wydania partii, utrzymanie integralności we wszystkich formatach zapisów jest niepodważalne. GMP wymaga tego samego poziomu integralności zarówno dla zapisów papierowych, jak i elektronicznych, chociaż konkretne kontrole mogą się różnić w zależności od formatu.
- Systemy papierowe: Najlepsze praktyki obejmują używanie kontrolowanych formularzy z niezmywalnym tuszem i jasnymi protokołami korekty (e.g. , poprawki przez przekreślenie pojedynczą linią z podpisami i datami). Bezpieczne archiwizowanie i przestrzeganie określonych okresów przechowywania są również kluczowe.
- Systemy elektroniczne: Powinny działać na zwalidowanym oprogramowaniu, które jest zgodne z Aneks 11 i GAMP 5 . Kluczowe funkcje obejmują dostęp oparty na rolach, unikalne uwierzytelnianie użytkowników, kompleksowe ścieżki audytu oraz zsynchronizowane znaczniki czasu. Regularne przeglądy ścieżek audytu są niezbędne do identyfikacji i rozwiązania wszelkich nieprawidłowości.
- Systemy hybrydowe: Te stanowią największe ryzyko, ponieważ obejmują zarówno zapisy elektroniczne, jak i papierowe. Na przykład, gdy instrument generuje dane elektroniczne, które są później przepisywane do papierowego dziennika, oryginalny elektroniczny wynik musi być zachowany jako główny zapis. Kroki uzgadniania powinny być wbudowane w przepływ pracy, aby wykrywać i rozwiązywać wszelkie rozbieżności między zapisami elektronicznymi a papierowymi. Jest to szczególnie istotne w produkcji mięsa hodowlanego, gdzie nawet drobne niespójności danych mogą zagrozić środkom kontroli zanieczyszczeń.
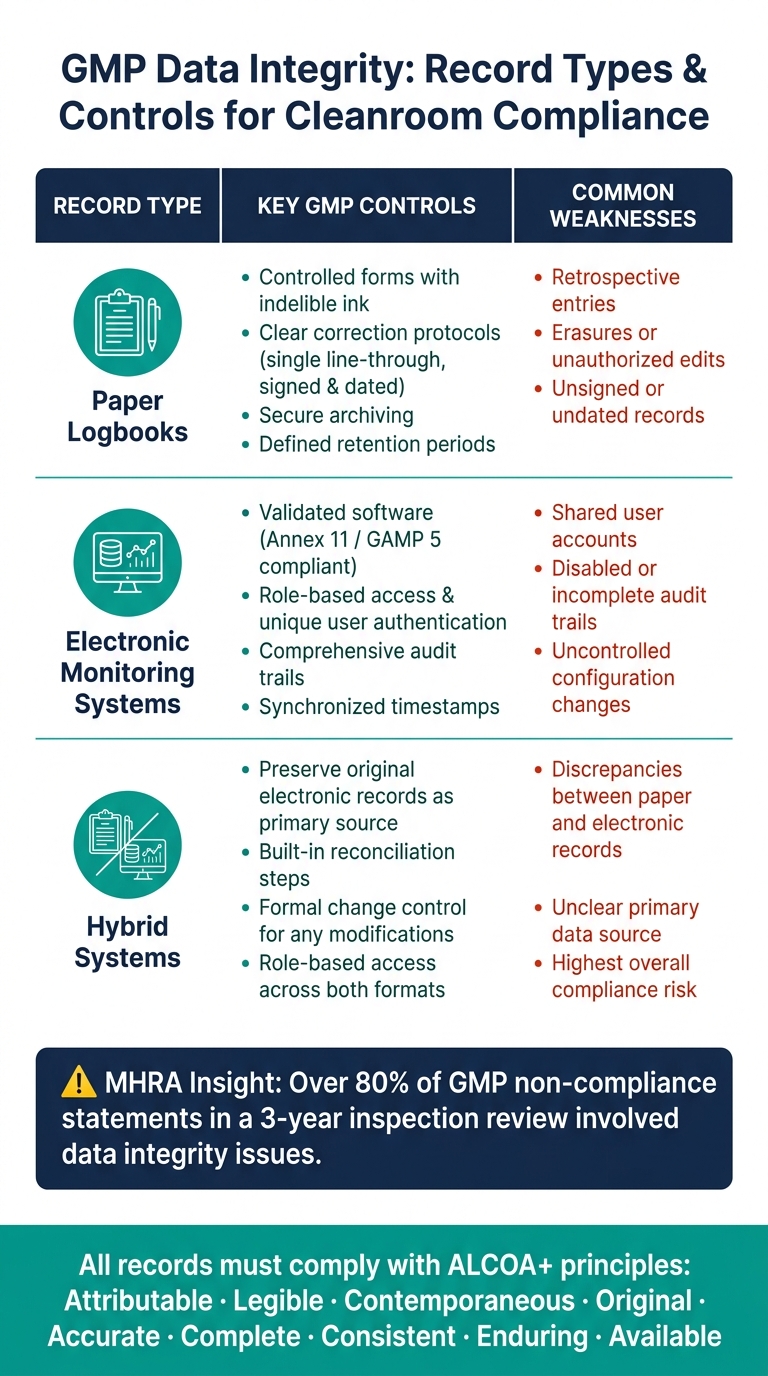
Poniższa tabela podsumowuje kluczowe kontrole i powszechne słabości dla każdego typu rekordu:
| Typ rekordu | Kluczowe kontrole GMP | Powszechne słabości |
|---|---|---|
| Kontrolowane formularze, niezmywalny atrament, jasne protokoły korekty, podpisane i datowane wpisy | Wpisy retrospektywne, wymazy, niepodpisane rekordy | |
| Elektroniczne systemy monitorowania | Zweryfikowane oprogramowanie, dostęp oparty na rolach, ścieżki audytu, synchronizacja czasu | Wspólne konta użytkowników, wyłączone lub niekompletne ścieżki audytu |
| Systemy hybrydowe | Zachowanie oryginalnych elektronicznych rekordów; wdrożenie kroków uzgadniania | Rozbieżności między papierowymi a elektronicznymi rekordami, niejasne źródło danych pierwotnych |
Aby zapewnić zgodność, zapisy powinny być kategoryzowane według ich krytyczności.Dla obiektów produkujących mięso hodowlane, dane związane z decyzjami o wypuszczeniu partii lub kontrolą zanieczyszczeń (e.g. , wyniki monitorowania środowiska, logi alarmów HVAC lub dane z testów integralności filtrów) powinny podlegać najsurowszym kontrolom dostępu, częstym przeglądom i solidnemu zarządzaniu ścieżką audytu.
sbb-itb-ffee270
Rutynowe monitorowanie środowiska w czystych pomieszczeniach GMP & Integralność danych 21CFR część 11
Zarządzanie cyklem życia danych w czystych pomieszczeniach
Integralność danych GMP: Rodzaje zapisów & Kontrole zgodności w czystych pomieszczeniach
Etapy cyklu życia danych w czystych pomieszczeniach
Cykl życia danych w czystych pomieszczeniach w obiektach produkujących mięso hodowlane obejmuje kilka etapów, z których każdy ma określone wymagania dotyczące zgodności.
Generowanie danych oznacza początek.Obejmuje to odczyty z instrumentów takich jak liczniki cząstek, czujniki różnicy ciśnień, sondy temperatury i wilgotności, próbki powietrza, płytki kontaktowe powierzchni oraz dzienniki weryfikacji czyszczenia. Dla każdego parametru musi być udokumentowana częstotliwość pobierania próbek, wyznaczony operator oraz skalibrowany instrument. Dopasowanie tych zadań monitorujących do etapów produkcji - takich jak inokulacja, ekspansja komórek czy zbiór - pomaga wykazać, jak kontrola środowiskowa bezpośrednio łączy się z jakością i bezpieczeństwem produktu.
Po wygenerowaniu, dane wchodzą w etap przechwytywania i transferu. Idealnie, systemy elektroniczne powinny automatycznie rejestrować odczyty z wpisami oznaczonymi czasem, powiązanymi z indywidualnymi kontami użytkowników. W przypadku wpisów papierowych, dane muszą być rejestrowane w czasie rzeczywistym przy użyciu niezmywalnego tuszu, z kontrolami uzgadniania podczas przenoszenia danych do systemów elektronicznych.
Faza przechowywania jest równie krytyczna.Zarówno surowe, jak i przetworzone dane muszą być zachowane, aby każda zgłoszona wartość mogła być prześledzona do swojego oryginalnego zapisu. Wymaga to bezpiecznych, zweryfikowanych repozytoriów z kontrolą dostępu opartą na rolach oraz regularnego testowania kopii zapasowych. Kopie zapasowe powinny być przechowywane w oddzielnej lokalizacji od systemu głównego i okresowo weryfikowane, aby zapewnić możliwość ich przywrócenia w razie potrzeby.
Na koniec, archiwizacja kończy cykl życia. Rekordy przechodzą w stan tylko do odczytu, z kontrolowanym dostępem, gdy nie są już aktywnie używane, ale muszą pozostać dostępne przez wymagany okres przechowywania. W zakładach produkujących mięso hodowlane, archiwizacja danych z fazy rozwoju może również wspierać przyszłe wysiłki walidacyjne.
Jasne zrozumienie tych etapów jest niezbędne do skutecznego zarządzania ryzykiem, jak opisano poniżej.
Typowe ryzyka w zarządzaniu danymi
Przetwarzanie danych podczas transferów stwarza znaczne ryzyko.Błędy w ręcznej transkrypcji i retrospektywne wpisy mogą podważać integralność danych. Aby tego uniknąć, wszystkie wpisy muszą być zgodne z zasadami ALCOA+ (Atrybutywność, Czytelność, Współczesność, Oryginalność, Dokładność, plus Kompletność, Spójność, Trwałość i Dostępność) w czasie rzeczywistym.
Zmiany konfiguracji są kolejnym poważnym problemem. Dostosowania limitów alarmowych, mapowania czujników lub ustawień zegara systemowego bez formalnej kontroli zmian mogą zagrozić wiarygodności danych zarejestrowanych przed i po zmianie. Dodatkowo, awarie przechowywania - czy to z powodu uszkodzonych baz danych, nieprzetestowanych kopii zapasowych, czy archiwów papierowych uszkodzonych przez czynniki środowiskowe - mogą sprawić, że krytyczne zapisy staną się niedostępne. Aby zminimalizować te ryzyka, upewnij się, że każdy strumień danych jest przypisany do wyznaczonego punktu archiwizacji z wyraźnym właścicielem, co zmniejsza prawdopodobieństwo wykrycia luk podczas inspekcji regulacyjnych.
Kontrole GMP dla systemów monitorowania pomieszczeń czystych
Krytyczne kontrole dla systemów monitorowania
System monitorowania spełniający standardy GMP opiera się na skalibrowanych czujnikach, bezpiecznym przetwarzaniu danych i efektywnym zarządzaniu alarmami. Czujniki dla parametrów takich jak temperatura, wilgotność względna, różnica ciśnień, liczba cząstek nieożywionych i próbki mikrobiologiczne muszą być kalibrowane zgodnie z udokumentowanymi harmonogramami i śledzone do uznanych standardów. Automatyzacja transferu danych z tych czujników, wraz z zsynchronizowanymi znacznikami czasu, minimalizuje ryzyko błędów manualnych.
Zarządzanie alarmami jest równie istotne. Limity alarmów powinny być zgodne z ramami regulacyjnymi, takimi jak ISO 14644-1 limity klas i wytyczne EU GMP Aneks 1. Każdy wyzwolony alarm musi być opatrzony zarejestrowaną odpowiedzią, w tym szczegółami użytkownika, znacznikami czasu i wszelkimi komentarzami.Nieudokumentowanie reakcji na alarm stwarza lukę w zgodności.
Kontrole dostępu oparte na rolach muszą być ściśle egzekwowane w całym systemie. Autoryzacja na poziomie administratora powinna być wymagana do wszelkich zmian limitów alarmów, konfiguracji czujników lub ustawień zegara systemowego, a te zmiany muszą podlegać formalnemu procesowi kontroli zmian. Ścieżki audytu są obowiązkowe dla wszystkich działań związanych z GMP, takich jak aktualizacje konfiguracji, usuwanie danych, podpisy elektroniczne i dostosowania czujników. Te ścieżki wymagają regularnych przeglądów, zgodnie z wytycznymi MHRA dotyczącymi integralności danych i załącznikiem 11 EU GMP.
Dla systemów produkcji mięsa hodowanego, te kontrole są szczególnie ważne, ponieważ warunki środowiskowe mają bezpośredni wpływ na żywotność komórek.
Gdy te kontrole są wdrożone, system musi zostać zwalidowany, a zmiany muszą być starannie zarządzane, aby zapewnić ciągłą zgodność.
Procedury Walidacji i Kontroli Zmian
Systemy monitorowania muszą przejść walidację poprzez etapy IQ, OQ i PQ, aby zweryfikować dokładność czujników, funkcjonalność alarmów, integralność danych, procesy tworzenia kopii zapasowych i ścieżki audytu. Alternatywnie można zastosować podejście cyklu życia zgodne z zasadami GAMP 5 i Aneksu 11.
Aneks 1 EU GMP (rewizja z 2022 roku) wymaga, aby systemy monitorowania środowiska były "odpowiednio kwalifikowane i walidowane" oraz nakazuje, aby elektroniczne zapisy spełniały standardy Aneksu 11 dotyczące integralności, bezpieczeństwa i śledzenia. Te wymagania stanowią podstawę dla każdej placówki zgodnej z GMP.
Wszelkie modyfikacje, które mogą wpłynąć na integralność danych, funkcjonalność alarmów lub śledzenie, muszą przejść formalny proces kontroli zmian. Nawet pozornie drobne aktualizacje, takie jak poprawki oprogramowania, mogą zakłócić ścieżki audytu i nie powinny być wdrażane bez wcześniejszej oceny wpływu.
Różne typy danych wymagają dostosowanych kontroli GMP, aby zapewnić dokładne i terminowe raportowanie.
Porównanie typów danych i wymagań zgodności
Każdy typ danych w monitorowaniu czystości pomieszczeń ma specyficzne wymagania, aby utrzymać zgodność z GMP.Tabela poniżej przedstawia kluczowe kontrole dla różnych typów danych:
| Typ danych | Tryb monitorowania | Kluczowe kontrole GMP | Podstawa limitu |
|---|---|---|---|
| Liczba cząstek nieżywotnych | Ciągłe lub częste podczas operacji w klasie A/B; rutynowe w innych klasach | Walidowane liczniki cząstek; automatyczne przechwytywanie danych; alarmy; kalibracja śledzona do standardów; ślad audytu dla zmian konfiguracji | Limity klasy ISO 14644-1; wytyczne załącznika 1 dla stref klasy A/B |
| Różnica ciśnień | Ciągłe; alarmy | Kalibrowane przetworniki ciśnienia; automatyczne, oznaczone czasem rejestrowanie; rejestrowane potwierdzenia alarmów; utrzymanie różnicy 10–15 Pa między strefami | Załącznik 1; projekt klasyfikacji pomieszczeń specyficzny dla obiektu |
| Temperatura i wilgotność względna | Ciągłe dla procesów krytycznych; okresowe w innych miejscach | Kalibrowane sondy; automatyczne zbieranie danych; analiza trendów; limity alarmowe oparte na potrzebach procesowych i regulacyjnych | Wiedza o procesie; wytyczne regulacyjne; wrażliwość produktu |
| Żywe mikroorganizmy w powietrzu | Okresowe (aktywne pobieranie próbek powietrza); zwiększona częstotliwość dla operacji krytycznych | Kwalifikowani próbobrani; kontrolowane procedury pobierania próbek; łańcuch dowodowy do laboratorium; wyniki powiązane z partią i lokalizacją; dokumentacja gotowa do dochodzenia | Limity mikrobiologiczne EU GMP Aneks 1 według klasy |
| Wyniki kontaktu powierzchniowego | Okresowe; po czyszczeniu i po operacji | Metody próbkowania kontrolowanego; śledzenie laboratoryjne; wyniki sprawdzane w odniesieniu do specyficznych limitów jakości; powiązane z zapisami czyszczenia | EU GMP Aneks 1; SOP obiektu |
Każdy typ danych wymaga zdefiniowanych kryteriów akceptacji, regularnych harmonogramów przeglądów, polityk przechowywania oraz procesu eskalacji dla odchyleń.Stosowanie jednolitych standardów przeglądu dla wszystkich typów danych jest powszechnym błędem, który regulatorzy coraz częściej poddają kontroli. Dostosowanie procesu przeglądu do specyficznych potrzeb każdego typu danych zapewnia zgodność i efektywność operacyjną.
Raportowanie, Przegląd i Działania Korygujące
Tworzenie Zgodnych Raportów
Raporty zgodne ze standardami ALCOA+ muszą być dokładne, precyzyjne i dostępne do audytów. Raport monitorowania czystego pomieszczenia zgodny z GMP powinien być zwięzły, weryfikowalny i zdolny do wspierania decyzji o zwolnieniu partii, jednocześnie demonstrując kontrolę środowiskową. Co najmniej, te raporty powinny:
- Obejmować okres monitorowania i zakres.
- Podsumować działania związane z pobieraniem próbek w porównaniu do planowanego harmonogramu.
- Wyraźnie stwierdzić, czy jakiekolwiek limity alarmowe lub działania zostały przekroczone.
Analiza trendów jest kluczowym elementem tych raportów, wykorzystującym narzędzia statystyczne, takie jak wykresy kontrolne, średnie kroczące i wskaźniki odchyleń na 100 próbek, aby zidentyfikować stopniowe zmiany. Na przykład, miesięczny trend pokazujący stały wzrost liczby żywych komórek w pobliżu linii zbioru bioreaktora dostarcza znacznie więcej informacji niż pojedyncze zdarzenie poza limitem. Dodanie adnotacji, takich jak działania konserwacyjne, dostosowania procesów lub zmiany personelu, ułatwia interpretację danych i przygotowanie do audytu.
Przeglądy ścieżki audytu są kolejnym kluczowym krokiem, wymagającym od przeszkolonego personelu dokładnego dokumentowania swoich ustaleń. Obejmuje to rejestrowanie, kto przeglądał konkretne zdarzenia systemowe, odnotowywanie wszelkich anomalii oraz szczegółowe opisywanie działań następczych, wszystko w podpisanym i datowanym zapisie.
Częstotliwość raportów powinna być dostosowana do związanego z nimi ryzyka. Na przykład:
- Raporty związane z partią są tworzone dla każdego cyklu produkcyjnego.
- Podsumowania rutynowego monitorowania środowiska są zazwyczaj tygodniowe lub miesięczne.
- Raporty trendów są przygotowywane miesięcznie lub kwartalnie w celu identyfikacji wczesnych oznak odchyleń.
Wybrana częstotliwość raportowania musi być uzasadniona w standardowych procedurach operacyjnych (SOP) i konsekwentnie przestrzegana. Te protokoły stanowią również podstawę do inicjowania działań korygujących, gdy zidentyfikowane zostaną odchylenia.
Rozwiązywanie problemów z zgodnością
Gdy występują odchylenia, niezbędna jest strukturalna, możliwa do śledzenia odpowiedź. Każde odchylenie powinno mieć unikalny identyfikator, jasny opis i ocenę ryzyka, która ocenia zarówno wpływ na produkt, jak i integralność danych. Odchylenia muszą być również sklasyfikowane (drobne, poważne lub krytyczne) i powiązane z konkretnymi partiami lub seriami produkcyjnymi, aby ocenić, czy wydanie partii jest zagrożone lub czy potrzebne są dodatkowe testy.
Ramy CAPA (Działania Korygujące i Zapobiegawcze) są kluczowe dla rozwiązywania problemów związanych z GMP. Skuteczne CAPA wymaga więcej niż przypisywania zdarzeń do "błędu ludzkiego." Wytyczne EMA i PIC/S podkreślają:
"niewystarczające badanie krytycznych odchyleń, wyników OOS i problemów z integralnością danych" jest częstą przyczyną działań egzekucyjnych.
Narzędzia analizy przyczyn źródłowych, takie jak 5 Why lub diagramy rybiej ości, są nieocenione w odkrywaniu problemów systemowych - niezależnie od tego, czy dotyczą one luk proceduralnych, niewystarczającego szkolenia, czy słabości w kontrolach technicznych. Działania korygujące powinny odnosić się do ryzyk zidentyfikowanych podczas gromadzenia i przechowywania danych.
Każde CAPA musi zawierać mierzalne kryteria skuteczności. Na przykład: "brak powtórzonych przekroczeń poziomu działania dla klasy B przez sześć miesięcy." Dodatkowo, przeglądy kontrolne są niezbędne, aby zapewnić spełnienie tych kryteriów.Typowe metryki CAPA obejmują:
- Liczba otwartych działań.
- Średni czas zamknięcia.
- Procent działań zakończonych na czas.
- Wskaźnik powtarzających się odchyleń, który jest silnym wskaźnikiem skuteczności CAPA.
Przegląd listów ostrzegawczych GMP z lat 2015-2019 ujawnił, że 65–70% cytatów dotyczących integralności danych wynikało z niewystarczających dochodzeń, brakującej dokumentacji lub nieprawidłowego przeglądu i raportowania danych [2]. To podkreśla znaczenie solidnego raportowania i responsywnego systemu CAPA jako dowodu na dobrze kontrolowany obiekt.
Utrzymanie zgodności z GMP w zakładach produkujących mięso hodowlane
Aby zapewnić bezpieczeństwo i jakość w produkcji mięsa hodowlanego, zakłady muszą dostosować ustalone kontrole GMP do specyficznych wyzwań tego nowo powstającego obszaru.Ponieważ integralność danych w pomieszczeniach czystych odgrywa kluczową rolę w utrzymaniu bezpieczeństwa produktów, udoskonalenie praktyk GMP dla mięsa hodowlanego jest niezbędne.
Dostosowanie Praktyk GMP dla Mięsa Hodowlanego
Ramowe zasady GMP, takie jak EU Annex 1, pierwotnie stworzone dla farmaceutyków, wymagają dostosowania, aby uwzględnić unikalne ryzyka w produkcji mięsa hodowlanego. Formalna ocena ryzyka, taka jak analiza w stylu FMEA lub HACCP, zapewnia solidną podstawę do dostosowania zasad GMP do każdego etapu produkcji. Krytyczne operacje, takie jak rozmrażanie banku komórek, inokulacja bioreaktora, ekspansja komórek i zbiór, wymagają odpowiednich klasyfikacji pomieszczeń czystych, protokołów ubierania się i monitorowania środowiska, zgodnie z wymaganiami Aneksu 1.Tymczasem zadania w dół strumienia, takie jak obsługa rusztowań i pakowanie, mogą przestrzegać standardów higieny GMP dla żywności zgodnie z Rozporządzeniem (WE) nr 852/2004, pod warunkiem, że śledzenie i integralność danych pozostają nienaruszone przez cały proces [6] [9][14].
Strategie monitorowania środowiska powinny koncentrować się na organizmach istotnych dla mięsa hodowlanego i bezpieczeństwa żywności, a nie tylko na tradycyjnych patogenach farmaceutycznych. Pobieranie próbek powinno być priorytetem w obszarach wysokiego ryzyka, takich jak te w pobliżu otwartych bioreaktorów, stref przygotowania mediów i stacji obsługi rusztowań. Te lokalizacje powinny być wybierane na podstawie udokumentowanych wzorców przepływu powietrza i analiz ruchu personelu [9][10].
Ze względu na dużą ilość danych generowanych przez bioreaktory mięsa hodowlanego, systemy muszą być zdolne do przechwytywania, oznaczania czasem i bezpiecznego przechowywania tych danych w zweryfikowanym repozytorium. Oryginalny surowy plik danych z instrumentu powinien zawsze być identyfikowany jako główny zapis, aby zapewnić zgodność [7][8].
Protokoły czyszczenia i dezynfekcji również wymagają starannego rozważenia. Pozostałości uznane za akceptowalne w środowiskach farmaceutycznych mogą zakłócać adhezję komórek lub różnicowanie w produkcji mięsa hodowlanego. Dane weryfikacyjne dla środków czyszczących powinny być zbierane i utrzymywane jako część programu monitorowania środowiska [3][4].
Korzystanie z zasobów branżowych, takich jak Cellbase
Specjalistyczne platformy zakupowe są nieocenione w zaspokajaniu specyficznych potrzeb zakładów produkujących mięso hodowlane. Sprzęt do czystych pomieszczeń zgodny z GMP powinien spełniać standardy ochrony przed wnikaniem i czystości, a także integrować się bezproblemowo z zatwierdzonymi systemami danych. Dostawcy muszą dostarczać szczegółowe specyfikacje, w tym możliwości śledzenia audytu, formaty eksportu danych, konfiguracje alarmów i procedury kalibracji, wraz ze sprzętem.
Podczas pozyskiwania systemów przez
- Funkcje integralności danych: bezpieczne ścieżki audytu, zapisy z oznaczeniem czasowym, uprawnienia oparte na rolach i unikalne loginy użytkowników
- Kompatybilność systemu: wsparcie dla standardowych protokołów komunikacyjnych i API do centralnego przechowywania danych
- Kalibracja i konserwacja: dostępność kompleksowej dokumentacji
- Wsparcie kwalifikacji: szablony IQ/OQ dostarczane przez dostawcę do usprawnienia walidacji
- Przydatność do pomieszczeń czystych: materiały i projekty ułatwiające czyszczenie
Wczesne żądanie dokumentacji od dostawcy w procesie zakupu może pomóc uniknąć potencjalnych problemów z kwalifikacją w przyszłości [3][4].
Wniosek
Podsumowanie kluczowych punktów
Zgodność z GMP w zarządzaniu danymi w pomieszczeniach czystych opiera się na wykazaniu kontroli - nad procesami, zapisami i decyzjami opartymi na tych danych. Jeśli zapis jest niewiarygodny, proces, który dokumentuje, staje się równie wątpliwy. Zasada ta ma zastosowanie uniwersalne, niezależnie od tego, czy dotyczy dzienników monitorowania środowiska, wyników bioreaktorów, raportów o odchyleniach, czy certyfikatów kalibracji.
W trakcie tej dyskusji wyłoniły się cztery główne tematy. Po pierwsze, integralność danych, kierowana zasadami ALCOA+ jest fundamentem zgodnej dokumentacji w pomieszczeniach czystych. Po drugie, zarządzanie cyklem życia zapewnia, że dane są rejestrowane dokładnie, przeglądane na czas, przechowywane bezpiecznie i zachowywane przez wymagany okres. Po trzecie, zwalidowane i kontrolowane zmiany systemy monitorowania stanowią techniczną podstawę, której żadna SOP nie może zastąpić.Jak podkreślono w analizie inspekcji GMP przeprowadzonych przez MHRA w latach 2016-2021, do najczęstszych niedociągnięć nadal należą niekompletne zapisy i niewystarczające przeglądy ścieżki audytu [1]. Wreszcie, dokładne i możliwe do prześledzenia raportowanie zapewnia, że surowe dane mogą być powiązane z decyzjami dotyczącymi partii, dochodzeniami i działaniami naprawczymi, spełniając oczekiwania regulacyjne.
Dla zakładów produkujących mięso hodowlane zasady te nabierają jeszcze większego znaczenia. Wyzwanie polegające na połączeniu przepływów pracy w stylu R&D z kontrolami na poziomie produkcji wymaga solidnego zarządzania danymi, aby połączyć oba środowiska operacyjne. Właściwe zarządzanie danymi w pomieszczeniach czystych nie tylko zapewnia spójność i powtarzalność, ale także przygotowuje zakłady do zwiększenia skali produkcji, jednocześnie demonstrując bezpieczeństwo produktu regulatorom, inwestorom i konsumentom.
Najbardziej praktyczna rada? Zajmij się znanymi ryzykami, zanim audytorzy je podkreślą.Luki, takie jak hybrydowe systemy papierowo-elektroniczne, współdzielone loginy użytkowników, opóźnione przeglądy danych i niekontrolowane lokalne przechowywanie, są przewidywalne i możliwe do zapobieżenia. Proaktywne rozwiązywanie tych problemów jest znacznie bardziej efektywne - i mniej kosztowne - niż odtwarzanie ścieżki danych po incydencie jakościowym.
Dla zespołów poszukujących sprzętu monitorującego, czujników lub infrastruktury dostosowanej do tych potrzeb,
FAQs
Jak można wykazać zasady ALCOA+ w codziennych zapisach z czystych pomieszczeń?
Aby zastosować zasady ALCOA+ w codziennych zapisach z czystych pomieszczeń, upewnij się, że:
- Przypisywalność: Wyraźnie zidentyfikuj osobę odpowiedzialną, w tym znaczniki czasu dla każdego wpisu.
- Czytelne : Rekordy muszą być łatwe do odczytania i wolne od dwuznaczności.
- Współczesne: Dokumentuj informacje w momencie, gdy aktywność ma miejsce.
- Oryginalne: Zachowaj pierwsze nagranie danych, nie kopie ani transkrypcje.
- Dokładne: Upewnij się, że wszystkie wpisy odzwierciedlają prawdziwe dane bez błędów.
- Kompletne: Zawieraj wszystkie istotne dane i metadane bez pominięć.
- Spójne: Zachowaj logiczny, sekwencyjny porządek w rekordach.
- Trwałe: Używaj formatów i materiałów odpowiednich do długoterminowego przechowywania.
- Dostępne: Przechowuj rekordy dostępne do przeglądu lub audytów, gdy są potrzebne.
Te kroki są kluczowe dla zapewnienia zgodności z Dobrą Praktyką Wytwarzania (GMP) w zarządzaniu danymi w pomieszczeniach czystych.
Jakie są główne zagrożenia dla integralności danych w hybrydowych systemach papierowo-elektronicznych?
Przechowywanie informacji w wielu lokalizacjach wprowadza komplikacje w weryfikacji dokładności danych. Dodatkowo, ręczne wprowadzanie danych zwiększa ryzyko błędów ludzkich, podczas gdy słabo kontrolowane lub samodzielne systemy pozostawiają zapisy podatne na manipulacje lub usunięcie. Te problemy podkreślają potrzebę silnych praktyk zarządzania danymi w celu utrzymania zgodności i zachowania integralności danych.
Jakich dowodów oczekują inspektorzy na walidację systemu i kontrolę zmian?
Inspektorzy często proszą o udokumentowane dowody pokazujące walidację systemu. Obejmuje to testowanie krytycznych parametrów, takich jak:
- Integralność filtra HEPA: Zapewnienie, że filtry spełniają wymagane standardy wydajności.
- Przepływ powietrza i różnice ciśnień: Weryfikacja, czy mieszczą się one w dopuszczalnych zakresach, aby utrzymać kontrolowane środowiska.
- Dane monitorowania środowiska: Pokazanie, że obiekt spełnia wymagania dotyczące czystości i kontroli zanieczyszczeń.
Poza testami walidacyjnymi, utrzymywanie dokumentacji działań związanych z kontrolą zmian jest równie ważne. Obejmuje to działania takie jak wymiany filtrów czy modyfikacje obiektu, które pomagają udowodnić, że system nadal działa zgodnie z oczekiwaniami i spełnia normy regulacyjne.










