

Si vous modifiez d'abord et vérifiez ensuite, vous pouvez corriger un changement hors cible dans le clone. Je garderais le flux de travail simple : choisissez la méthode d'édition la moins risquée, limitez l'exposition à l'éditeur, puis testez à la fois les sites hors cible prévus et la stabilité du clone avant la libération.
Pour les ingénieurs en bioprocédés, les scientifiques en culture cellulaire et les équipes R&D de viande cultivée, le point principal est simple. Les systèmes CRISPR peuvent encore couper à des sites presque correspondants, souvent avec 3–6 mésappariements tolérés, et ces erreurs peuvent se transmettre dans des clones monocellulaires étendus. L'article divise le contrôle des risques en trois phases : avant l'édition, pendant l'édition, et après l'édition.
Voici la liste complète en termes simples :
-
Choisissez l'outil d'édition à faible risque pour le travail
- Utilisez l'édition de base ou l'édition prime lorsqu'elles peuvent effectuer la modification sans rupture double brin
- Utilisez la modulation basée sur dCas9 si vous avez seulement besoin de régulation génique
- Si vous avez besoin d'une nucléase, commencez par une variante Cas9 à haute fidélité
-
Sécurisez le matériel de départ
- Confirmez l'identité de la lignée cellulaire
- Vérifiez la mycoplasme
- Enregistrez le numéro de passage
- Séquencez le locus cible réel dans la lignée de travail, pas seulement le génome de référence
-
Sélectionnez les guides avant le travail humide
- Utilisez des outils basés sur l'alignement et des outils basés sur le scoring pour les cibles hors site ensemble
- Préférez un guide avec un profil hors cible plus propre plutôt qu'un guide avec seulement une activité sur cible plus élevée
- Surveillez la longueur du guide, 40–60% de contenu en GC, et les séquences homopolymères
-
Limitez l'exposition à l'intérieur de la cellule
- Utilisez RNP ou la livraison d'ARNm au lieu de systèmes plasmidiques ou viraux lorsque cela est possible
- Utilisez la dose efficace minimale
- Évitez de prolonger la persistance de l'éditeur juste pour forcer les résultats de transfection
-
Ajoutez des contrôles supplémentaires pour les cas à risque plus élevé
- Envisagez des nickases appariés
- Utilisez des systèmes , Cas9 scindé, inductibles ou contrôlés par la lumière lorsque le timing est important
- Ajoutez des protéines anti-CRISPR comme étape d'arrêt si nécessaire
-
Valider correctement après modification
- Confirmer d'abord la modification sur cible
- Vérifier chaque site hors cible prédit avec NGS d'amplicon ciblé
- Passer à GUIDE-seq, CIRCLE-seq, CAST-seq, UDiTaS, ou WGS lorsque le risque du projet est plus élevé
- Pour les éditeurs de base ou les éditeurs primaires , ajouter des vérifications au niveau de l'ARN lorsque pertinent
-
Ne pas libérer un seul clone sur la seule séquence
- Comparer 2–3 clones indépendants
- Utiliser un contrôle parental non modifié
- Supprimer les clones avec instabilité ou dérive phénotypique
- Libérer uniquement lorsque l'état de modification, le dépistage hors cible et les enregistrements sont tous complets
Une façon courte d'y penser : concevoir pour éviter les coupures hors cible, livrer pour limiter le temps dans la cellule, puis valider à la profondeur que le risque du projet justifie. C'est le fil conducteur de toute la pièce.
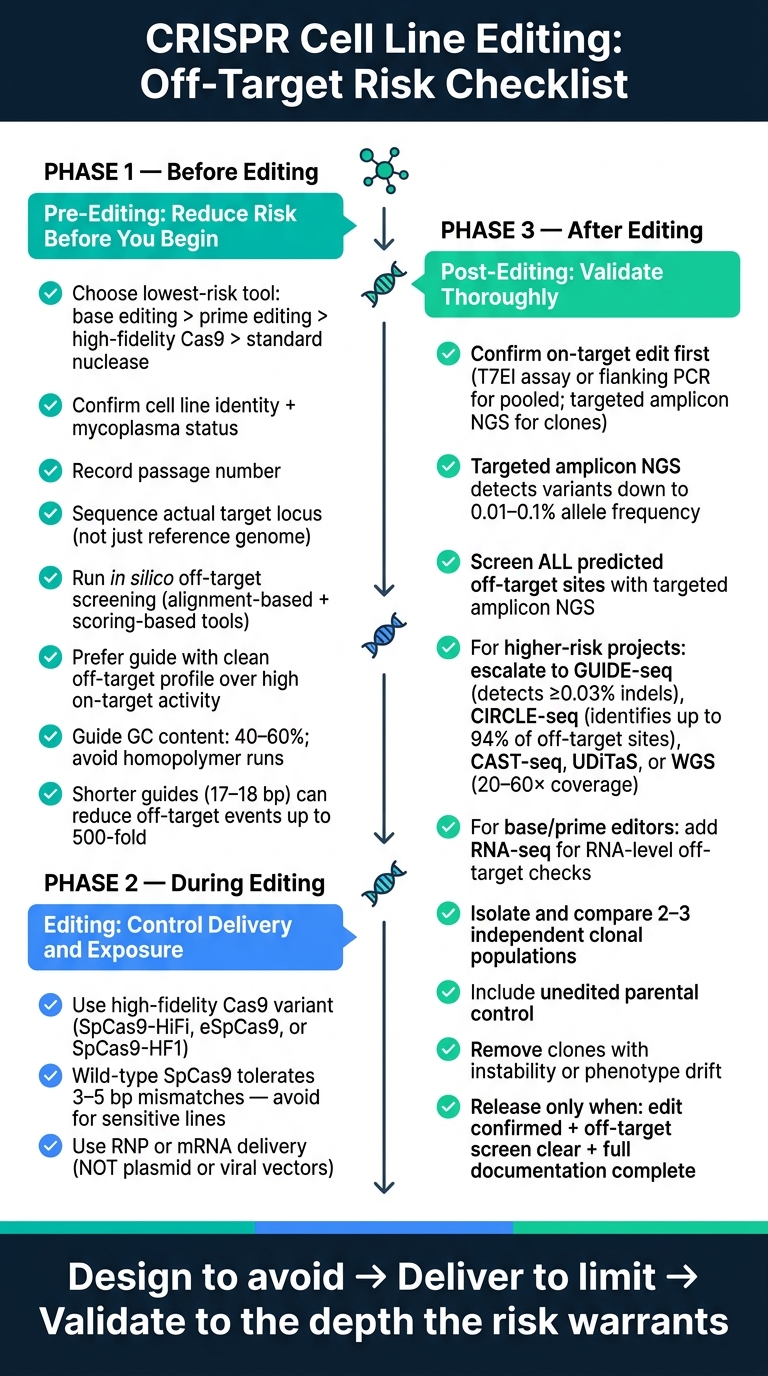
Contrôle du risque hors cible CRISPR : Liste de contrôle en 3 phases pour l'édition de lignées cellulaires
Liste de contrôle avant édition : réduire le risque avant le début de l'édition
Définir l'objectif de l'édition et choisir la méthode d'édition la moins risquée
Avant de commander un seul réactif, soyez très clair sur ce que l'édition est censée accomplir. Un knockout, un knock-in, un changement de nucléotide unique et une modulation transcriptionnelle ne portent pas le même risque hors cible. Ils n'exigent pas non plus le même outil.
L'ordre de risque général est simple. Les nucléases formant des DSB telles que Cas9 et Cas12 se situent à l'extrémité supérieure du risque car elles peuvent entraîner de grandes délétions, des translocations et des réponses aux dommages de l'ADN [1] [7]. Les éditeurs de base et les éditeurs principaux utilisent des nickases, évitant ainsi les DSBs et réduisant le risque de variation structurelle [1][5]. Pour la modulation transcriptionnelle, les éditeurs épigénétiques tels que dCas9 fusionné à des modificateurs transcriptionnels laissent la séquence d'ADN inchangée [1].
La règle pratique est simple : utilisez la méthode la moins génotoxique qui peut encore fournir la modification dont vous avez besoin. Pour les changements de nucléotides uniques, les CBEs ou ABEs conviennent mieux que le HDR, qui peut encore introduire des indels [3] [1]. Pour les substitutions et les petites insertions ou délétions, l'édition principale montre souvent une activité hors cible plus faible que le CRISPR-Cas9 standard [1]. Si vous devez utiliser une nucléase, choisissez une variante à haute fidélité telle que SpCas9-HiFi, eSpCas9, ou SpCas9-HF1 [1] [6].
Une fois l'approche d'édition définie, verrouillez la lignée cellulaire de travail et la séquence cible exacte.
Confirmez l'identité de la lignée cellulaire, son historique et la séquence du locus cible
Si la lignée cellulaire est mal identifiée ou contaminée, le reste du flux de travail commence à vaciller. Même un ARN guide bien conçu ne sauvera pas un mauvais matériel de départ. Vérifiez l'identité de la lignée cellulaire avant de commencer toute édition. En même temps, confirmez le statut de mycoplasme et enregistrez le numéro de passage actuel, car les cellules à passage élevé peuvent modifier la stabilité génomique et l'efficacité de l'édition [1][6].
Tout aussi important, ne vous fiez pas uniquement à un génome de référence. Séquencez le locus cible exact dans la lignée cellulaire de travail. Cette étape vous aide à repérer les SNPs ou indels qui peuvent bloquer la liaison du guide ou créer de nouveaux sites hors cible [1] [6].
Après cela, passez à la conception du guide.
Effectuez un dépistage in silico des sites hors cible avant de sélectionner les réactifs
Une fois le locus cible confirmé, dépistez les ARN guides candidats in silico avant de vous engager dans le travail de laboratoire humide. Utilisez à la fois des outils basés sur l'alignement, tels que Cas-OFFinder ou FlashFry , et des outils basés sur le scoring, tels que le scoring CFD ou DeepCRISPR. Le premier groupe aide à trouver des sites génomiques avec une homologie de séquence. Le second aide à classer ces sites par probabilité de clivage prédite [1][5].
Lors de la présélection des guides, un profil hors-cible plus propre devrait surpasser l'efficacité brute sur cible. Un guide avec une efficacité sur cible de 70% et sans prévisions de hors-cibles est un point de départ plus sûr qu'un guide avec 90% d'efficacité et plusieurs sites à haut risque [6]. Dans certains contextes, raccourcir la longueur du guide de 20 pb à 17-18 pb peut réduire les événements hors-cible jusqu'à 500 fois sans perte significative de précision sur cible [5]. Visez une teneur en GC entre 40% et 60%, et évitez les séquences de quatre bases identiques ou plus [6][5].
Cela dit, le criblage in silico a ses limites. Il ne prend pas bien en compte l'état de la chromatine, le cycle cellulaire ou le contexte spécifique à la cellule [1][6][4]. Considérez-le comme un filtre, pas une preuve. Cela restreint le champ, mais cela ne remplace pas la confirmation expérimentale.
Avancez les sites prédits à haut risque dans le plan d'édition et de validation.
sbb-itb-ffee270
Liste de contrôle d'édition : choix de l'éditeur, livraison et exposition
Utilisez des éditeurs à haute spécificité et des ARN guides bien classés
Commencez par la liste restreinte des hors-cibles prédits et utilisez-la pour choisir l'éditeur. Dans la plupart des cas, une variante SpCas9 à haute fidélité - SpCas9-HiFi, eSpCas9, ou SpCas9-HF1 - est un meilleur choix par défaut que le SpCas9 de type sauvage [6] [1]. Le SpCas9 de type sauvage peut tolérer jusqu'à trois à cinq mésappariements de paires de bases, en particulier dans la région distale du PAM, et cela crée un risque hors-cible significatif dans les lignées cellulaires sensibles [3].
Une règle simple aide ici : utilisez l'éditeur haute fidélité le moins actif qui réalise toujours la modification souhaitée.
Pour les éditeurs de base, suivez les modifications collatérales et les effets hors cible de l'ARN séparément du risque hors cible de l'ADN [1] [8]. Ce sont des modes de défaillance différents, et ils nécessitent des vérifications séparées. Si vous pouvez effectuer la modification sans cassures double brin, l'édition de base ou l'édition prime peut mieux convenir dans des flux de travail à risque plus élevé [1][8].
Une fois l'éditeur choisi, la prochaine tâche est de réduire au maximum son temps à l'intérieur de la cellule.
Limitez la persistance de l'éditeur avec une livraison transitoire et une dose efficace minimale
La persistance de l'éditeur est tout aussi importante que le choix de l'éditeur.Plus l'éditeur reste actif dans la cellule, plus il a de temps pour agir sur des sites à faible probabilité. Cela fait du format de livraison un point de contrôle majeur.
Utilisez une livraison transitoire telle que les RNPs ou l'ARNm, et évitez l'ADN plasmidique ou les vecteurs viraux qui prolongent l'expression de l'éditeur [1] [5]. En pratique, la livraison par RNP devrait être la norme [6] .
La dose compte aussi. Une concentration élevée de nucléase augmente la probabilité de clivage sur des sites hors cible à faible sensibilité [5]. Utilisez la dose efficace minimale. Si l'efficacité de la transfection est faible, ne vous contentez pas d'ajouter plus de réactif en espérant le meilleur. Cela déplace souvent le problème au lieu de le résoudre.
Ajouter des mesures de protection de précision pour les flux de travail à risque plus élevé
Certaines procédures nécessitent des garde-fous supplémentaires. C'est particulièrement vrai pour les cibles proches des oncogènes, suppresseurs de tumeurs, ou dans les lignées cellulaires sensibles au p53, où un événement hors cible peut avoir un coût disproportionné [1][6][3].
Les mesures de protection utiles incluent :
- Nickases appariées, qui nécessitent deux coupures proches. Une seule coupure hors cible est généralement réparée sans mutation, donc le risque hors cible diminue considérablement par rapport à une configuration de nucléase standard [4][1].
- Systèmes Cas9 induisibles, contrôlés par la lumière ou divisés, qui aident à maintenir l'activité de l'éditeur dans une fenêtre étroite lorsque la livraison est efficace et que l'exposition doit rester brève [1].
- Protéines Anti-CRISPR (Acr), qui agissent comme un interrupteur d'arrêt. Ces protéines Acr naturellement présentes peuvent désactiver le complexe CRISPR-Cas après un intervalle défini, vous offrant un frein moléculaire sur l'activité de l'éditeur [1].
Liste de contrôle post-édition : détecter les événements hors cible et valider les clones
Analyser les sites hors cible prédits avec un séquençage ciblé
Une fois l'édition terminée, confirmez d'abord le changement prévu au locus cible. Pour un premier passage rapide dans des cellules groupées, vous pouvez utiliser un test de clivage des mésappariements tel que T7 Endonuclease I, une digestion par restriction, ou une PCR de flanquement.Soyez prudent avec l'interprétation : chacune de ces méthodes a des limites de sensibilité, surtout pour les modifications rares ou les variantes homozygotes [9].
Pour la validation au niveau du clone, le NGS d'amplicon ciblé est la norme. Il vous offre une vue quantitative de la fréquence allélique et peut détecter des variantes jusqu'à 0,01 % à 0,1 % [3].
Séquencez chaque site hors cible prédit avec le NGS d'amplicon ciblé. Cela devrait être l'étape de validation par défaut.
Escaladez vers des essais à l'échelle du génome ou structurels lorsque le risque du projet est plus élevé
Le dépistage site par site n'est pas toujours suffisant. Si l'éditeur, le locus cible ou la lignée cellulaire suggère un risque caché, passez à des essais capables de détecter des événements que vous n'aviez pas prévus à l'avance.
Les essais de découverte à l'échelle du génome tels que GUIDE-seq et CIRCLE-seq n'ont pas besoin de listes de sites hors cible préalables. GUIDE-seq peut détecter des sites hors cible avec des fréquences d'indels aussi basses que 0,03% [2]. CIRCLE-seq peut identifier jusqu'à 94% des sites hors cible in vitro [3]. Ces méthodes sont utiles lorsque le contexte du type cellulaire peut masquer l'activité hors cible.
Si vous êtes préoccupé par les réarrangements importants, les lectures d'amplicons standard peuvent manquer le principal problème. Les délétions, inversions et translocations nécessitent des tests conçus pour les changements structurels, tels que CAST-seq et UDiTaS [1].
Le séquençage du génome entier (WGS) est l'option la plus large. Il peut détecter les indels, les variations structurelles et les changements de nombre de copies à travers le génome [1]. Le compromis est la profondeur et le coût : il nécessite généralement une couverture de 20–60×, ce qui en fait un mauvais choix pour le dépistage de routine des populations en vrac [1].
Utilisez NGS d'amplicons ciblés pour les sites prédits. Passez à des essais à l'échelle du génome ou structurels pour les projets à risque plus élevé. Pour les éditeurs de base ou de prime, ajoutez l'ARN-seq pour vérifier les effets hors cible au niveau de l'ARN.
Sélectionnez plusieurs clones indépendants et documentez les critères de libération
Après les vérifications de séquence, testez le phénotype dans plus d'un clone.
Ne pas avancer avec un seul clone édité. Isolez et développez au moins deux à trois populations clonales indépendantes et comparez-les avec un contrôle parental non édité [4] [9]. Retirez les clones qui montrent une instabilité ou une dérive phénotypique [3]. Ensuite, confirmez la modification prévue à l'état allélique requis, qu'il soit hétérozygote ou homozygote, en utilisant le séquençage ciblé par amplicon NGS [9].
La documentation n'est pas un travail administratif à la fin. Elle fait partie de la libération du clone. Enregistrez le fond génétique de la lignée parentale, la conception de l'ARNsg, la variante de la nucléase, la méthode de livraison, et tous les résultats de QC [3]. Un clone ne doit avancer que lorsque la modification prévue est confirmée, les sites hors cible prévus sont clairs, et l'enregistrement complet est en place.
Édition du génome avec CRISPR: Comment minimiser efficacement les effets hors cible
Conclusion : une liste de contrôle en trois phases pour des modifications de lignées cellulaires plus propres
Ensemble, la liste de contrôle traite le contrôle hors cible comme un processus en plusieurs étapes, et non comme une vérification de QC ponctuelle. L'objectif est simple : réduire le risque tôt, limiter l'activité de l'éditeur, puis vérifier le résultat.
La profondeur de validation doit correspondre au risque. Ne libérez que des clones indépendants multiples confirmés à l'état allélique prévu.
FAQs
Pourquoi ne pas se fier à un seul clone ?
Se fier à un seul clone est risqué. L'édition CRISPR n'est pas parfaitement spécifique, et peut donc introduire des mutations hors cible non intentionnelles.
C'est pourquoi les équipes développent généralement plusieurs populations clonales. Cela facilite la recherche d'une ligne qui porte l'édition cible souhaitée sans modifications hors cible nuisibles.
Il y a une autre raison aussi : les lignées cellulaires peuvent montrer une hétérogénéité génétique. Séquencer plusieurs clones aide à confirmer que le knockout homozygote ou autre modification du site cible est présent à travers les loci cibles.
Quand l'amplicon NGS est-il suffisant ?
Le séquençage de nouvelle génération basé sur l'amplicon est souvent suffisant lorsque vous avez besoin d'une méthode ciblée et rentable pour confirmer les sites potentiellement hors cible signalés par des outils informatiques ou d'autres méthodes de dépistage.
Le séquençage du génome entier reste le seul moyen de quantifier pleinement les effets hors cible. Mais pour de nombreuses applications, ce niveau d'analyse n'est tout simplement pas nécessaire.
Comment choisir l'éditeur le plus sûr ?
Choisissez la variante de nucléase CRISPR la moins active qui coupe encore bien votre site cible.
Vous ne pouvez pas choisir la meilleure variante uniquement à partir de la prédiction. La seule façon fiable est de réaliser un petit dépistage à travers des variantes de nucléase sélectionnées et de lire l'édition avec le séquençage de nouvelle génération.
Pour la viande cultivée R&D, cela vous offre une voie pratique à suivre : commencez par une courte liste de variantes, puis testez les plus faibles étape par étape jusqu'à ce que vous trouviez l'option la moins active qui édite toujours le site cible efficacement.









