

Les dossiers de lot sont essentiels pour la conformité et la sécurité des produits. Ils documentent chaque étape de la production, garantissant que les normes réglementaires sont respectées. Pour les producteurs de viande cultivée, maintenir la stérilité et des dossiers détaillés est non négociable. Les inspections de la FDA soulignent souvent des problèmes tels que des données manquantes, des examens incomplets et de mauvaises actions correctives, ce qui peut entraîner des avertissements ou des perturbations.
Points Clés:
- Dossiers de Lot: Deux types - Dossier de Lot Maître (MBR) (la "recette") et Dossier de Production de Lot (BPR) (l'"exécution").
- Problèmes Courants: Erreurs humaines (50% des problèmes), contrôles en cours de processus manquants, examens incomplets et mauvais systèmes CAPA (Action Corrective et Préventive).
- Normes de la FDA: Le respect des principes ALCOA+ (Attribuable, Lisible, Contemporain, Original, Précis, Complet, Cohérent, Durable, Disponible) est obligatoire.
- Solutions: Les audits indépendants, les dossiers électroniques de lots et la vérification rigoureuse des fournisseurs peuvent minimiser les erreurs et améliorer la conformité.
Les entreprises de viande cultivée comme UPSIDE Foods ont établi une référence en assurant une documentation détaillée, la traçabilité des intrants et des mesures correctives rapides. En s'inspirant de ces pratiques, les producteurs peuvent éviter les écueils réglementaires et maintenir des normes de haute qualité.
Guide complet de la documentation et de la tenue de registres pour la conformité FDA dans les sciences de la vie
sbb-itb-ffee270
Problèmes courants dans la documentation des dossiers de lots
Les rapports d'inspection de la FDA soulignent systématiquement un problème récurrent : les écarts dans l'examen des dossiers de production figurent parmi les principales déficiences GMP citées par les régulateurs [7]. Pour les producteurs de viande cultivée, ces lacunes vont au-delà de simples erreurs administratives - elles compromettent la capacité à démontrer des conditions stériles soutenues. Ces problèmes apparaissent sous plusieurs formes, comme illustré dans les exemples ci-dessous.
Examens Incomplets et Non-Conformités
Un problème courant est l'échec des unités de contrôle qualité à examiner minutieusement les dossiers de lots. Au lieu de faire partie intégrante du processus de libération, les examens se produisent souvent de manière réactive - seulement après qu'un problème de produit ait déjà émergé [7]. Cette approche laisse des lacunes significatives dans les dossiers de production.
Par exemple, Davis City Pharmacy a reçu une observation FDA 483 en raison de dossiers de lots manquant de détails critiques tels que les quantités de composants, les étapes opérationnelles et les initiales du personnel. De même, CAPS a été cité pour absence de signatures requises et de vérifications des examinateurs dans les entrées clés [3]. Ces manquements ne sont pas des incidents isolés ; des études montrent qu'environ 52 % des violations de documentation s'aggravent en l'absence de systèmes de gestion bioprocessus robustes bioprocess management systems [3].
"Ce n'est pas la complexité du processus qui déclenche des citations - c'est l'incohérence, l'incomplétude et le manque de supervision." - Services de conseil en audit GXP & Consulting Services [5]
Absence de registres de vérification en cours de processus
Une autre déficience fréquente est l'absence de documentation appropriée pour les vérifications en cours de processus. Ces registres sont cruciaux, surtout aux points de contrôle critiques dans les opérations aseptiques. Par exemple, Nephron Sterile Compounding Center a fait l'objet de citations pour ne pas avoir documenté les étapes essentielles de l'habillage et les procédures aseptiques dans leurs dossiers de lot [3]. Pour les producteurs de viande cultivée, où la stérilité est primordiale, de telles omissions rendent impossible la confirmation de la conformité aux mesures de contrôle de la contamination.
Amphastar a également été signalé pour ne pas avoir enquêté ou documenté les variations de rendement inattendues ou les écarts de production [3]. Les risques de telles négligences sont flagrants. Dans un cas, une installation pharmaceutique non identifiée en 2024/2025 a été trouvée stockant des dossiers de lots complétés sur des étagères ouvertes et des bureaux. Les enquêteurs de la FDA ont découvert des pages manquantes, y compris sept d'un seul dossier, et une section entière "Solution de Synthèse" absente d'un autre [6].
Échecs de CAPA et de Vérification GMP des Fournisseurs
Au-delà des erreurs de documentation, l'absence de processus correctifs efficaces et de vérification des fournisseurs compromet davantage la fiabilité des dossiers de lots. Lorsqu'il y a des écarts de production sans rapport de Corrective and Preventive Action (CAPA) correspondant, l'intégrité des dossiers de lots est compromise [7]. Par exemple, Eugia Pharma Specialities Limited, inspecté entre le 22 janvier et le 2 février 2024, a reçu un FDA 483 pour ne pas avoir examiné les écarts de manière adéquate. Leur système CAPA inefficace et leurs enquêtes incomplètes ont conduit à des problèmes de production répétés, forçant une refonte complète de leurs procédures d'enquête et de CAPA [9].
De même, lors d'une inspection du 26 septembre au 25 octobre 2023, Stokes Healthcare Inc. a démontré une mauvaise gestion des écarts. L'entreprise n'a pas étendu les enquêtes à tous les lots affectés et a retardé la finalisation de leurs analyses [9].
"Un écart sans rapport CAPA ou de déviation correspondant ? C'est un échec de conformité." - Services de conseil en audit GXP & [5]
Les problèmes de vérification des fournisseurs ajoutent une autre couche de complexité. Empower Clinic Services LLC a été cité lors d'une inspection du 18 juillet au 5 août 2022 pour des procédures de contrôle qualité inadéquates, y compris des qualifications de fournisseurs insuffisantes et des processus d'enquête médiocres [9]. Pour les producteurs de viande cultivée, qui dépendent des milieux de culture, des lignées cellulaires et d'autres intrants critiques, garantir la conformité GMP des fournisseurs est essentiel pour maintenir l'intégrité des dossiers de lots.
Exigences de la FDA pour les dossiers de lots
Les règles de la FDA pour les dossiers de lots tournent autour de 21 CFR Part 117, qui établit la base pour la sécurité alimentaire.Lorsqu'il s'agit de viande cultivée, où le maintien de la stérilité pendant la phase de culture cellulaire est crucial, la documentation doit souvent répondre aux normes plus strictes de Partie 111 ou Partie 211, en plus de la Partie 117 [10][14]. Cela souligne à quel point une documentation précise est essentielle pour garantir la sécurité et l'efficacité de la production de viande cultivée.
Normes de base pour les dossiers de lot
Chaque lot nécessite deux documents clés :
- Dossier de lot maître (MBR): Le modèle approuvé décrivant le processus de production.
- Dossier de production de lot (BPR): Un enregistrement détaillé de ce qui se passe réellement pendant le cycle de production [12][2].
Le BPR doit inclure des détails tels que les numéros de lot ou de série, les détails de l'équipement, les dates de nettoyage, les identifiants des composants, les mesures exactes et les comparaisons des rendements réels par rapport aux rendements théoriques [10][14].
"Le dossier de production par lot doit suivre avec précision le dossier de fabrication maître approprié et vous devez effectuer chaque étape de la production du lot." – 21 CFR 111.255 [12]
Chaque étape critique doit être enregistrée immédiatement, avec les initiales de l'exécutant et du vérificateur notées [10][11]. La FDA exige le respect des principes ALCOA(+), ce qui signifie que les dossiers doivent être Attribuables, Lisibles, Contemporains, Originaux et Précis - ainsi que Complets, Cohérents, Durables et Disponibles [1].
Si une déviation par rapport au Dossier de Fabrication Maître se produit, elle doit être examinée en profondeur. Cela inclut la documentation du problème, la réalisation d'une analyse des causes profondes et la mise en œuvre d'un plan d'Action Corrective et Préventive (CAPA) [8] [1]. Les évaluations initiales des déviations doivent être enregistrées dans les 24 à 48 heures suivant la détection [8]. Pour les installations utilisant des systèmes électroniques, la conformité avec 21 CFR Part 11 est obligatoire. Cela inclut des signatures électroniques validées et des pistes d'audit sécurisées et horodatées [8] [1].
Procédures de Conservation et de Révision des Enregistrements
Des processus appropriés de conservation et de révision des enregistrements sont essentiels pour rester conforme et garantir la sécurité des produits.Dans la production stérile, comme celle de la viande cultivée, chaque détail dans les dossiers de lot doit faire l'objet d'un examen minutieux. L'équipe Contrôle Qualité (QC) est responsable de l'examen de tous les dossiers de lot, de la surveillance des résultats et des données de test avant qu'un lot puisse être approuvé pour la distribution [10] [13].
"Tous les dossiers de production et de contrôle des produits médicamenteux doivent être examinés et approuvés par l'unité de contrôle qualité avant qu'un lot ne soit libéré ou distribué." – 21 CFR 211.192 [2]
Les fabricants visent souvent à compléter 95 % des examens de lot dans les 30 jours suivant la production [2] . Cependant, pour les processus stériles plus complexes impliqués dans la viande cultivée, les examens prennent généralement 7 à 10 jours, avec des installations à haute performance atteignant des délais inférieurs à 7 jours [2]. Les systèmes d'enregistrement électronique par lots peuvent accélérer considérablement ces examens, tels que ceux intégrés dans les systèmes de production de viande cultivée, - réduisant le temps de moitié par rapport aux méthodes sur papier - tant qu'ils sont validés pour répondre aux exigences de la Partie 11 et maintenir l'intégrité des données [1].
Ce que les entreprises de viande cultivée approuvées par la FDA ont bien fait
Les entreprises de viande cultivée approuvées par la FDA ont placé la barre haute en adoptant des pratiques qui répondent aux défis de la documentation et respectent des normes de sécurité rigoureuses.
Lorsque UPSIDE Foods est devenue la première entreprise de viande cultivée à passer la consultation pré-commercialisation de la FDA en novembre 2022, elle a établi un modèle pour l'industrie.La FDA a émis une lettre de "pas d'autres questions" après avoir examiné en profondeur leur processus de production, qui comprenait l'établissement de lignées cellulaires, les banques cellulaires, les contrôles de fabrication, et tous les composants et intrants [16]. Cette réalisation a souligné l'importance d'une documentation détaillée pour répondre aux exigences strictes de la FDA.
Respect des normes de stérilité et de conformité
L'accomplissement remarquable d'UPSIDE Foods était leur approche exhaustive de la traçabilité des intrants. Chaque composant de production était soigneusement documenté, assurant une chaîne de responsabilité claire depuis la lignée cellulaire initiale jusqu'au produit final [16]. Ce niveau de transparence a permis aux examinateurs de la FDA de retracer chaque étape du processus de fabrication, confirmant que toutes les normes de sécurité étaient constamment respectées.
"La consultation préalable à la mise sur le marché de la FDA avec l'entreprise comprenait une évaluation du processus de production de l'entreprise et du matériel cellulaire cultivé fabriqué par le processus de production, y compris l'établissement de lignes cellulaires primaires et immortalisées et de banques cellulaires, les contrôles de fabrication, et tous les composants et intrants." – U.S. Food and Drug Administration [16]
D'autres entreprises prospères ont suivi l'exemple en mettant en œuvre une documentation détaillée des processus aseptiques. Cela comprenait des étapes critiques telles que les procédures d'habillage et les opérations de manipulation stérile [3]. Contrairement aux échecs de documentation antérieurs, ces entreprises ont employé des systèmes de révision par niveaux, impliquant des vérifications par les opérateurs, la supervision de la production et des examens par l'unité de qualité, pour détecter les erreurs potentielles avant la libération des lots [15]. Les systèmes d'enregistrement électronique par lot ont également joué un rôle crucial, en imposant des validations obligatoires à chaque étape et en maintenant des pistes d'audit immuables conformément aux exigences de la norme 21 CFR Part 11 [3][2].
Ces pratiques rigoureuses se sont naturellement étendues à la manière dont les entreprises géraient les écarts et les échecs.
Processus CAPA pour les échecs de lot
Lorsque les lots ne répondaient pas aux spécifications, les entreprises approuvées par la FDA prenaient des mesures rapides et systématiques. Leurs processus d'Action Corrective et Préventive (CAPA) comprenaient des analyses formelles des causes profondes, des évaluations d'impact et des actions correctives clairement documentées [3]. Tous les écarts étaient gérés dans un cadre intégré d'assurance qualité, garantissant que tous les problèmes étaient minutieusement investigués, justifiés et documentés avant que la production ne reprenne [2].
À l'avenir, l'intégrité des données devrait être un axe majeur des actions d'application de la FDA pour 2024–2025 [1].
Comment améliorer vos pratiques de gestion des dossiers de lot
Renforcer les pratiques de gestion des dossiers de lot nécessite une documentation précise pour aborder les échecs courants souvent identifiés lors des inspections de la FDA. Voici quelques stratégies pour relever les principaux défis.
Effectuer des audits indépendants des dossiers de lot
Des audits réguliers par des tiers peuvent révéler des problèmes que les examens internes pourraient négliger. Commencez par vous concentrer sur des systèmes critiques tels que les systèmes de gestion de l'information de laboratoire (LIMS), les systèmes d'exécution de la fabrication (MES) et la planification des ressources d'entreprise (ERP). Priorisez les documents ayant un impact réglementaire élevé, tels que les dossiers de tests de libération, les données de stabilité et les dossiers de production de lot.
Une méthode efficace est le test de récupération d'échantillons.Sélectionnez aléatoirement des lots récents et reconstituez leur historique de production et de laboratoire. Cela peut aider à identifier les données manquantes, les signatures incomplètes ou les lacunes dans la documentation qui pourraient entraîner des citations réglementaires. Vérifiez les pistes d'audit générées par le système avec les entrées manuelles pour identifier les modifications ou suppressions non autorisées.
Examinez tous les rapports Hors Spécifications (OOS) et Hors Tendance (OOT) de l'année passée. Évaluez si les analyses des causes profondes ont été approfondies et si les Actions Correctives et Préventives (CAPA) ont été correctement mises en œuvre. Il convient de noter que les problèmes de documentation représentent 21 % des lettres d'avertissement de la FDA, tandis que l'erreur humaine contribue à 50 % des problèmes de dossiers de lots dans la fabrication pharmaceutique [2].
"Ce n'est pas la complexité du processus qui déclenche des citations - c'est l'incohérence, l'incomplétude et le manque de supervision." – GXP Auditing & Services de Conseil [5]
Simulez des inspections réglementaires à travers des revues simulées périodiques. Cette pratique aide les équipes à reconnaître les incohérences et les problèmes potentiels d'intégrité des données avant un audit réel. Assurez-vous que tous les enregistrements suivent les principes ALCOA+ : Attribuable, Lisible, Contemporain, Original, Précis, Complet, Cohérent, Durable et Disponible.
Une fois l'intégrité de la documentation assurée, concentrez-vous sur la vérification de la qualité de tous les intrants de production.
Testez Tous les Intrants pour la Contamination Microbiologique
Des tests indépendants de stérilité et de puissance pour tous les intrants sont essentiels - ne vous fiez pas uniquement aux certificats d'analyse (CoAs) des fournisseurs. Cela est particulièrement crucial pour les producteurs de viande cultivée, car la contamination peut compromettre des lots entiers.
Par exemple, en février 2013, Central Admixture Pharmacy Services a fait face à des citations de la FDA en raison d'un contrôle microbien inadéquat lors de la libération de lots de produits stériles. L'entreprise a dû introduire des procédures détaillées de contrôle microbien dans ses Procédures Opératoires Standard (SOPs) [4].
Les points de contrôle microbiens en cours de processus peuvent prévenir une dépendance excessive aux tests de produit final. Intégrez ces points de contrôle dans les SOPs de libération de lots et maintenez une documentation stricte et contemporaine. Enregistrez tous les résultats des tests et les étapes de fabrication au fur et à mesure pour éviter les antidatations ou les entrées retardées, ce qui pourrait entraîner des citations de la FDA.
Conservez des dossiers complets des fournisseurs, y compris les CoAs, les rapports d'audit, les accords de qualité et un historique de toute déviation liée aux matériaux entrants.
Renforcer les pratiques d'enregistrement des lots implique également d'aligner les processus avec des normes établies comme le HACCP et le GCCP.
Aligner les dossiers avec les normes HACCP et GCCP
L'intégration des principes de l'Analyse des Dangers et Points Critiques pour leur Maîtrise (HACCP) dans les dossiers de lots garantit que les variables critiques du processus sont surveillées et documentées tout au long de la production. Cela inclut l'établissement de points de contrôle pour les tests microbiologiques en cours de processus plutôt que de se fier uniquement aux tests de la phase finale.
Pour les producteurs de viande cultivée, le respect des normes de Bonnes Pratiques de Culture Cellulaire (GCCP) est tout aussi essentiel. Les dossiers de lots doivent inclure des détails sur les manipulations aseptiques, les procédures d'habillage et la surveillance de l'environnement liés aux critères de libération des lots [3][4]. Ces étapes aident à maintenir la conformité et à assurer la sécurité du produit.
Les données de l'industrie montrent que 52 % des violations de documentation s'aggravent lorsque le logiciel de fabrication par lots approprié n'est pas en place [3][4]. Un exemple concret : en février 2023, le Nephron Sterile Compounding Centre a reçu une observation de la FDA en raison de l'absence de procédures de contrôle pour vérifier les variables critiques du processus avant la libération du lot [4]. Cela souligne la nécessité d'une documentation proactive alignée sur les normes reconnues.
Passer aux dossiers de lots électroniques (EBR) peut réduire considérablement les erreurs de documentation - jusqu'à 50% - grâce à la collecte de données en temps réel et aux flux de travail automatisés [2]. Ces systèmes signalent les résultats de tests microbiologiques manquants ou les examens incomplets avant qu'un lot n'avance, minimisant ainsi l'erreur humaine.
"La FDA s'attend à ce que les dossiers soient ALCOA(+): Attribuables, Lisibles, Contemporains, Originaux, Précis - plus Complets, Cohérents, Durables et Disponibles." – Atlas Compliance [1]
Toute divergence ou déviation inexpliquée dans les dossiers de lot doit être liée à une enquête formelle et à un système CAPA. Limitez les autorisations d'écriture et de suppression pour protéger l'intégrité des données de test microbiologiques électroniques. Les fabricants compétitifs visent à examiner et à libérer 95 % des lots dans les 30 jours suivant l'achèvement de la production [2].
Ces actions réduisent non seulement le risque de citations, mais s'alignent également sur les normes de documentation rigoureuses mises en évidence lors des récentes inspections de la FDA.
Biopharma vs Cultivated Meat: Différences dans les Dossiers de Lot
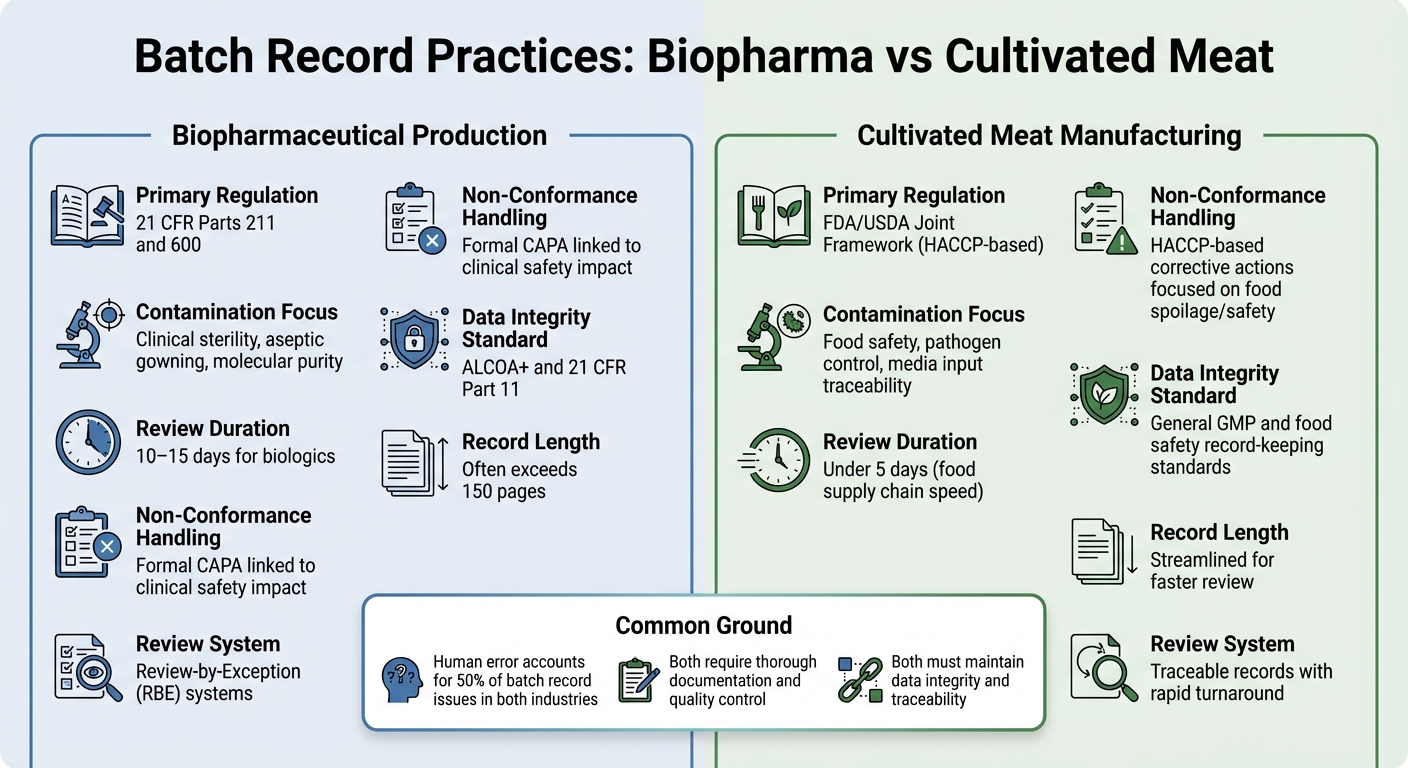
Comparaison des Exigences des Dossiers de Lot entre Biopharmaceutique et Viande Cultivée
Examiner les différences dans les pratiques de tenue des dossiers de lot entre la production biopharmaceutique et la fabrication de viande cultivée offre une image plus claire de la façon dont les exigences réglementaires façonnent les priorités de documentation dans ces industries.
Les deux secteurs nécessitent une documentation approfondie, mais leurs cadres réglementaires et objectifs de contrôle diffèrent considérablement. Dans le biopharma, les dossiers de lot sont strictement réglementés sous 21 CFR Parts 211 and 600, qui exigent que l'unité de Contrôle Qualité examine et approuve tous les dossiers de production et de contrôle avant qu'un lot puisse être libéré [2]. Les producteurs de viande cultivée, en revanche, suivent généralement les normes HACCP et GCCP.Ces éléments se concentrent davantage sur la sécurité alimentaire et le contrôle des agents pathogènes, plutôt que sur la stérilité de qualité clinique exigée pour les produits biologiques injectables.
Les dossiers de lots biopharmaceutiques sont souvent volumineux, dépassant parfois 150 pages, et le processus de révision peut prendre 10 à 15 jours. Pour rationaliser cela, de nombreuses entreprises biopharmaceutiques utilisent des systèmes Review-by-Exception (RBE), qui résument les principales déviations dans un rapport compact. Pendant ce temps, les producteurs de viande cultivée visent des dossiers traçables pouvant être examinés en moins de cinq jours, reflétant le rythme plus rapide de la chaîne d'approvisionnement alimentaire [2].
Le contenu de ces dossiers met également en évidence les priorités différentes. Les inspections biopharmaceutiques se concentrent souvent sur les détails du traitement aseptique, tels que les procédures d'habillage et les contrôles environnementaux. En revanche, les dossiers de viande cultivée doivent mettre l'accent sur les intrants de milieu et les tests microbiologiques pour garantir la sécurité alimentaire.Pour la viande cultivée, le défi réside dans le suivi des intrants médiatiques complexes, la documentation des tests microbiologiques pour tous les matériaux, et le respect des limites critiques de sécurité alimentaire - sans se conformer aux exigences de stérilité plus strictes des produits pharmaceutiques.
Tendances de contamination et de non-conformité
| Caractéristique | Production biopharmaceutique | Fabrication de viande cultivée |
|---|---|---|
| Réglementation principale | 21 CFR Parties 211 et 600 [2] | Cadre conjoint FDA/USDA (basé sur HACCP) |
| Focus sur la contamination | Stérilité clinique, habillage aseptique, pureté moléculaire [2] | Sécurité alimentaire, contrôle des pathogènes, traçabilité des intrants médiatiques |
| Durée de l'examen | 10–15 jours pour les produits biologiques [2] | Moins de 5 jours (vitesse de la chaîne d'approvisionnement alimentaire) |
| Gestion de la non-conformité | CAPA formel lié à l'impact sur la sécurité clinique [2] | Actions correctives basées sur le HACCP axées sur la détérioration/sécurité alimentaire |
| Norme d'intégrité des données | ALCOA+ et 21 CFR Part 11 [1] | Normes générales de tenue de registres GMP et de sécurité alimentaire |
Bien que les taux d'erreur humaine soient similaires dans les deux industries - environ 50 % des problèmes de dossiers de lots proviennent d'erreurs humaines [2] - les enjeux sont différents.Dans la biopharmacie, même une seule déviation non documentée pourrait avoir des implications graves pour la sécurité des patients. Pour la viande cultivée, les risques de contamination concernent davantage les agents pathogènes d'origine alimentaire et la détérioration, ce qui peut affecter l'ensemble des lots de production.
Conclusion
Les dossiers de lot servent de registre officiel pour chaque lot de production de viande cultivée - si une étape n'est pas enregistrée, les régulateurs considèrent qu'elle n'a pas été effectuée [6][3]. Cela souligne l'importance d'une documentation précise et d'un contrôle qualité strict.
Les inspections de la FDA soulignent que l'intégrité des données doit être conforme aux principes ALCOA+ [1]. Les équipes de contrôle qualité doivent examiner et approuver tous les dossiers de production avant qu'un lot puisse être libéré [2][17], et toute déviation doit être rapidement investiguée avec une analyse de la cause racine documentée [2][5]. Bien que l'erreur humaine représente 50 % des problèmes de dossiers de lots, les examens à deux niveaux et les processus CAPA (Corrective and Preventive Action) structurés peuvent aider à réduire ces risques [2][5].
"Ce n'est pas la complexité du processus qui déclenche les citations - c'est l'incohérence, l'incomplétude et la mauvaise supervision." - GXP Auditing & Services de conseil [5]
Pour surmonter ces défis, les producteurs de viande cultivée devraient se concentrer sur des audits indépendants, des tests rigoureux des ingrédients sûrs pour l'alimentation pour la contamination microbiologique, et s'assurer que la documentation respecte les normes HACCP et GCCP. La mise en œuvre de systèmes d'enregistrement électronique des lots, validés selon la partie 11 du 21 CFR [1], peut réduire considérablement les erreurs et accélérer les processus de révision.
L'environnement réglementaire exige de la précision, mais il est navigable. En apprenant des erreurs de la biopharma - telles que les signatures manquantes chez Qinhuangdao Zizhu Pharmaceutical [17], la vérification double insuffisante chez Terumo Corp [18] , et la documentation des écarts inadéquate chez Torrent Pharmaceuticals [18] - les entreprises de viande cultivée peuvent établir des systèmes conformes dès le départ. L'intégration de ces leçons permet une conformité proactive et une qualité constante. La conservation sécurisée des dossiers, le rapport des écarts en temps opportun et la réalisation d'audits simulés réalistes garantiront que les dossiers de lots restent prêts pour l'inspection et que les séries de production sont entièrement traçables.
Pour plus de ressources et de conseils d'experts sur le maintien de normes de production élevées dans la fabrication de viande cultivée, visitez
FAQs
Que doit inclure un dossier de lot pour la viande cultivée ?
Un dossier de lot pour la viande cultivée sert de journal complet de l'ensemble du processus de fabrication. Il doit inclure des instructions de traitement détaillées, des enregistrements d'exécution étape par étape, et noter toute déviation qui se produit pendant la production. De plus, il doit documenter les tests en cours de processus et les tests de libération pour confirmer que le produit répond aux normes de sécurité, de qualité et réglementaires.
Comment pouvons-nous prouver la stérilité à l'aide des dossiers de lot ?
Prouver la stérilité à travers les dossiers de lot implique d'examiner minutieusement les procédures de stérilisation documentées, les résultats des tests et les rapports de contrôle de qualité des milieux pour s'assurer qu'ils répondent aux exigences réglementaires.Il est crucial de traiter toute déviation ou tout test échoué par le biais d'enquêtes détaillées et de CAPA (Actions Correctives et Préventives). Ce processus garantit que chaque étape a été respectée et que tout problème a été correctement résolu pour maintenir les normes de stérilité.
Quand les dossiers de lots électroniques (Partie 11) sont-ils requis ?
Les dossiers de lots électroniques sont essentiels en vertu de la Partie 11 lorsque des systèmes électroniques sont utilisés pour documenter, enquêter et justifier les déviations des dossiers de lots. Ils jouent un rôle crucial pour garantir la conformité avec 21 CFR Part 211.192 , en protégeant l'intégrité des données, en respectant les délais d'enquête et en assurant une supervision efficace de la gestion.









